Article Text

Abstract
Background Characterisation of colorectal cancer (CRC) genomes by next-generation sequencing has led to the discovery of novel recurrently mutated genes. Nevertheless, genomic data has not yet been used for CRC prognostication.
Objective To identify recurrent somatic mutations with prognostic significance in patients with CRC.
Method Exome sequencing was performed to identify somatic mutations in tumour tissues of 22 patients with CRC, followed by validation of 187 recurrent and pathway-related genes using targeted capture sequencing in additional 160 cases.
Results Seven significantly mutated genes, including four reported (APC, TP53, KRAS and SMAD4) and three novel recurrently mutated genes (CDH10, FAT4 and DOCK2), exhibited high mutation prevalence (6–14% for novel cancer genes) and higher-than-expected number of non-silent mutations in our CRC cohort. For prognostication, a five-gene-signature (CDH10, COL6A3, SMAD4, TMEM132D, VCAN) was devised, in which mutation(s) in one or more of these genes was significantly associated with better overall survival independent of tumor-node-metastasis (TNM) staging. The median survival time was 80.4 months in the mutant group versus 42.4 months in the wild type group (p=0.0051). The prognostic significance of this signature was successfully verified using the data set from the Cancer Genome Atlas study.
Conclusions The application of next-generation sequencing has led to the identification of three novel significantly mutated genes in CRC and a mutation signature that predicts survival outcomes for stratifying patients with CRC independent of TNM staging.
- COLONIC NEOPLASMS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Large-scale mutational analyses by Sanger and next-generation sequencing has identified a handful of recurrently mutated genes in patients with colorectal cancer (CRC) mainly of Caucasian origin.
-
Prognostication of patients with CRC mainly relies on tumor-node-metastasis (TNM) staging.
-
BRAF mutation and microsatellite stability statuses also predict survival of patients with CRC.
What are the new findings?
-
Three novel recurrently mutated genes, namely CDH10, FAT4 and DOCK2, were identified to exhibit high mutation prevalence in the Asian CRC cohort.
-
The mutation status of a five-gene-signature (CDH10, COL6A3, SMAD4, TMEM132D, VCAN) could predict survival of patients with CRC independent of TNM staging in two independent cohorts.
How might it impact on clinical practice in the foreseeable future?
-
Identification of novel recurrently mutated genes will expand the current list of druggable targets and thus facilitate the development of new targeted therapeutics.
-
The five-gene-signature will help to stratify patients with early-stage CRC with different predicted clinical outcomes.
Introduction
Colorectal cancer (CRC) is the third most common cancer and the fourth leading cause of cancer-related death globally, and its incidence has been increasing rapidly in some areas of the world, including Asia.1 ,2 The molecular pathogenesis of CRC is characterised by successive acquisition of genetic alterations that lead to aberrant activation of proto-oncogenes and inactivation of tumour-suppressor genes. According to the classical tumour progression model of sporadic CRC proposed by Fearon and Vogelstein, APC mutation is involved in adenoma formation followed by oncogenic mutation of KRAS that promotes the transition from intermediate adenomas to carcinomas with TP53 inactivation as a late event.3 Since the last decade, research efforts have shifted from investigation of mutations of individual genes (eg, SMAD4) to genome-wide identification of genetic abnormalities in cancer.4 ,5 Sjöblom et al and Wood et al first used large-scale PCR-based sequencing to depict the genomic landscape of CRC, in which a number of well-known, high-frequency mutated genes identified as ‘gene mountains’ (ie, APC, KRAS, TP53, FBXW7) were found to be interspersed with many ‘gene hills’ that are mutated at low frequency.6 ,7 Leveraging the next-generation sequencing technology, The Cancer Genome Atlas (TCGA) Network reported the common occurrence of mutations in additional genes, such as ARID1A, SOX9 and FAM123B.8 These studies also demonstrate that CRC is highly genetically heterogeneous at the population level.
Identification of somatic mutations is key to understanding the molecular mechanism of CRC and the development of novel therapeutics. It is also presumed that genomic data could be used for disease prognostication to stratify patients with CRC with different clinical outcomes since mutations of specific genes are known to correlate with distinct biological behaviours of tumours.9 While adjuvant therapy is recommended for stage III patients with CRC, such treatment remains controversial for stage II patients because its toxicities may outweigh its benefits.10 It is therefore pivotal to stratify patients with CRC with dissimilar predicted outcomes for different treatment regimens. To date, prognostication of patients with CRC still heavily relies on tumor-node-metastasis (TNM) staging or similar histoclinical systems.11 Nevertheless, clinical outcomes of patients with the same histoclinical staging could be heterogeneous. Endeavours have been put forth to develop novel biological markers to make up for this insufficiency. To this end, microsatellite instability (MSI) resulting from defects in DNA mismatch repair is associated with better prognosis.12 BRAF mutation is also known to be associated with shortened survival in patients with late-stage CRC.13 Molecular profiling, such as gene expression patterns, has also been found useful for predicting clinical outcomes in CRC.14 However, the relationship between somatic mutation patterns at genome-wide level and clinicopathological features, including patients’ survival, in CRC has not yet been thoroughly investigated.
In the present study, we adopted a two-phase approach for genomic discovery in patients with CRC to identify potential novel recurrently mutated genes and mutation markers/patterns of prognostic value. We first performed exome sequencing to identify somatic mutations in 22 tumour tissues. Targeted capture sequencing of 187 recurrent and pathway-related genes in 160 CRC cases with detailed clinicopathological information was then conducted to evaluate their mutation prevalence and clinical relevance.
Methods
Sample collection and genomic DNA preparation
Genomic DNA was extracted from primary CRC tissues and matched lymphocyte samples using QIAamp DNA Mini Kit (Qiagen, Germany) and Gentra Puregene Blood Kit (Gentra Systems, Minneapolis, Minnesota, USA), respectively. All samples were collected from patients diagnosed with primary CRC without chemotherapy prior to surgery. After surgery, all stage I patients did not receive further chemotherapy whereas stage III and some stage II patients were treated with the 5-fluorouracil, leucovorin, and oxaliplatin (FOLFOX) regimen. Stage IV patients received either FOLFOX or 5-fluorouracil, folinic acid, irinotecan (FOLFIRI) regimen with irinotecan plus cetuximab as second-line treatment.
Illumina based whole-exome sequencing and reads alignment
Our bioinformatic pipeline is illustrated in figure 1. Genomic DNA from tumours and lymophocytes was fragmented and hybridised to commercially available capture arrays for enrichment. The exome capture procedure was performed with Agilent's SureSelect Human All Exon Kit protocol (Agilent Technologies). Resulting DNA libraries with an insert size of 200 bp on average were sequenced using the 90-bp paired-end technology on Illumina HiSeq 2000. Real-time image analysis and base calling were performed by Hiseq Control Software V.1.1.37 and Real Time Analysis V.1.7.45 using standard parameters, respectively. Before aligning reads to the Homo sapiens reference genome, we removed low quality reads that meet the following criteria: (1) reads include sequencing adaptors; (2) the ratio of ambiguous bases to read length ≥0.1; (3) read with more than five ambiguous bases. The resultant reads were aligned to reference genome hg18 by using BWA v0.5.9 (bwa aln -o 1 -e 50 -m 100 000 -t 4 -i 15 -q 10 -I).15 SAMtools was used to convert the SAM-formatted alignment results to BAM-formatted alignment files, followed by Genome Analysis Toolkit (GATK IndelRealigner) to calibrate alignment accuracy in local regions and Picard to mark duplicates.16 ,17
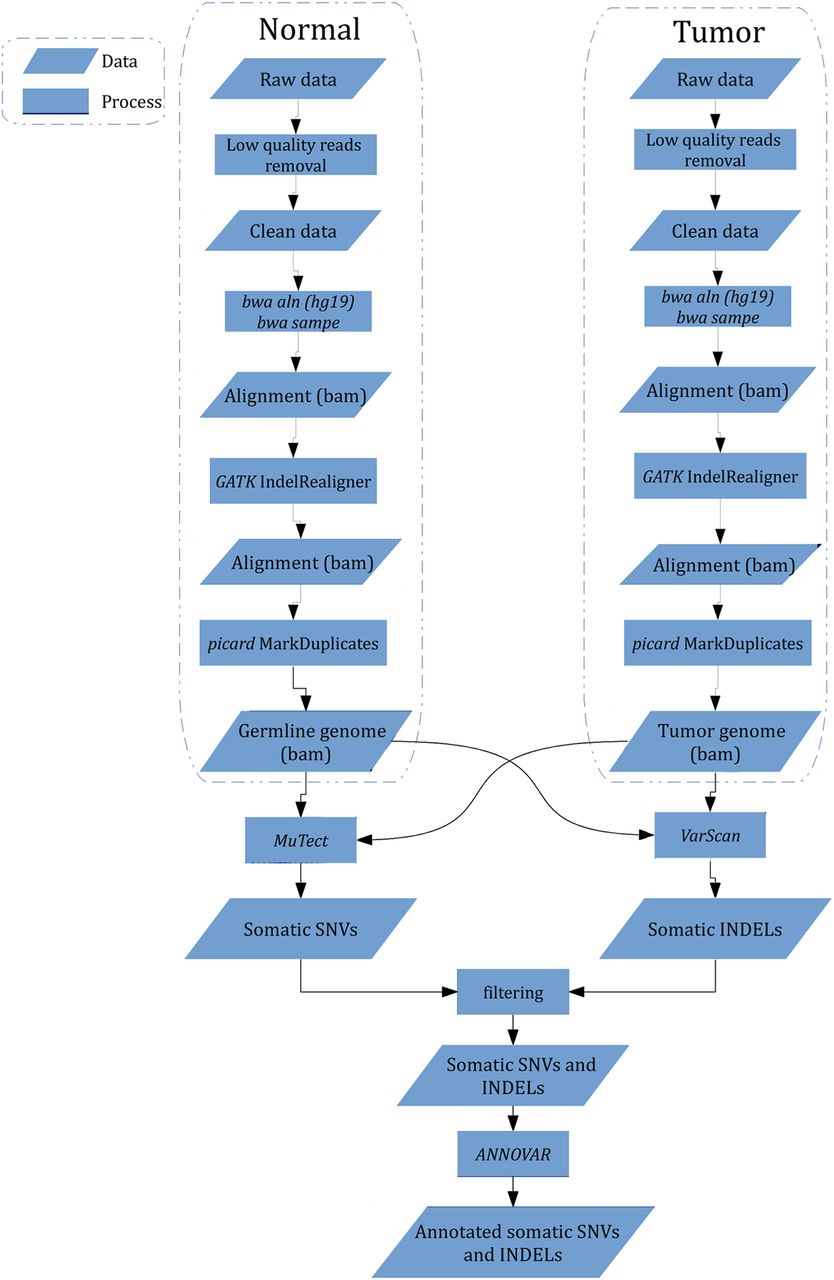
Work flow of identification of somatic single-nucleotide variation (SNV) and insertion and deletion (INDEL) from raw sequencing data. Low quality reads were removed, and bwa was used to perform alignment followed by alignment calibration by GATK and marked duplicates by picard. MuTect and VarScan were employed to detect somatic SNV and INDEL, processed by further filtering to eliminate false positives, respectively. All mutations were annotated with ANNOVAR.
Detection of somatic mutations and indels
MuTect was used to detect somatic mutations in discovery and validation cohorts, which is a sensitive tool to detect somatic point mutations, addressing tumour impurity and heterogeneity.18 After manual inspection, mutations found to be located in regions enriched for ‘mismatches’ were discarded. The minimum coverage was set at 10X, mutation allele fraction ≥10% and ≥5 reads that support this mutation. These somatic mutations were annotated with ANNOVAR.19 VarScan2 was used to detect somatic indels by comparing tumour BAM file against its matched normal BAM file with following parameters: min-coverage 10; min-coverage-normal 10; min-coverage-tumour 10; min-var-freq 0.1; min-freq-for-hom 0.75; somatic-p-value 0.05; min-avg-qual 0; Q 0.20 False-positive indels were removed through manual inspection. Significantly mutated genes (SMGs) were identified by MutSigCV.
Identification of significantly mutated pathways (SMPs)
SMPs are causally implicated in tumorigenesis and therefore their composing genes exhibit a higher-than-expected variant count due to selective advantages conferred by driver mutations. A statistical method as reported by Kan et al with modifications was employed to account for accuracy and computational speed.21
Statistical analysis
Relative risks of death associated with mutation(s) in the five-gene signature and other predictor variables were estimated from univariate Cox proportional hazards model first. Multivariate Cox models were also constructed to estimate the HR for mutation(s) in the five-gene signature. Overall survival in relation to mutation status was evaluated by the Kaplan-Meier survival curve and the log-rank test. Patients with more than 400-day follow-up survival data from TCGA study were used as an independent cohort for verification of prognostic significance of the five-gene signature. All analyses were performed using open source R software for Linux, V.2.15 (http://www.r-project.org/). A p value of less than 0.05 was taken as statistical significance. For driver gene prediction, q-value of less than 0.1 as used by other studies was considered statistically significant.22 ,23
Results
Identification of somatic mutations by exome sequencing in a discovery cohort of 22 patients with CRC
To delineate somatic mutations in patients with CRC, tumour and blood lymphocyte DNA of 21 cases were subject to whole-exome capture and massively parallel sequencing. Whole-genome sequencing was also performed in another CRC case, in which somatic mutations in exomic region were subject to further analysis. All 22 cases were microsatellite-stable (MSS) or with low MSI. Shotgun paired-end reads of 72-to-90-base-pair were generated and aligned onto human reference genome (UCSC hg18), resulting in a median haploid exome coverage of 57-fold and 49-fold from 22 CRC genomes and 22 matched lymphocyte controls, respectively (see online supplementary figure S1). At the sequencing depth of 50-fold, it has been estimated that >85% of somatic mutations with minimum variant allele frequency of 10% could be detected.18 As an independent-platform validation, 22 out of 25 (88.0%) somatic mutations identified by exome sequencing were successfully confirmed by Sanger sequencing. An average of 85.7% of exomic regions in CRC and 85.2% in lymphocyte samples were covered with more than 10 reads and were used for variant calling. The variant sets obtained from tumours were compared with matched lymphocyte DNA and dbSNP132 to establish the catalogue of cancer-specific, non-germline mutations in each tumour sample. A total of 1307 (996 non-silent and 311 silent mutations) somatic mutations were identified in the exomic regions of the 22 Chinese patients with CRC. The number of somatic mutations in 22 patients with CRC ranged from 13 to 109 with a median of 52.5 (see online supplementary table S1), which was not significantly different from that of non-hypermutated CRC reported by TCGA (ie, 58 per tumour; Wilcoxon test, p=0.52).8 Figure 2A illustrates that mutational changes observed in the 22 patients with CRC were predominated by C/G>T/A transition (49.1%). The number of mutations and the pattern of nucleotide changes were consistent with previous CRC genomics studies.6–8 The mutational landscape of these 22 patients with CRC was illustrated in figure 2B.

Identification of somatic mutations by exome sequencing in 22 patients with CRC for genomic discovery. (A) Spectrum of nucleotide alterations was determined in each exome-sequenced patient with CRC. Nucleotide change was predominated by C/G>T/A transition. (B) The landscape of non-silent mutations at genome-wide scale was depicted with the height of each gene reflecting the mutation frequency among 22 patients with CRC. Three reported gene mountains (ie, APC, KRAS, TP53) were interspersed with many novel gene hills (eg, FAT4, NF1, DOCK2, HERC2) discovered in our CRC cohort.
Recurrently mutated genes and altered pathways in the discovery cohort
Identification of recurrently mutated genes is key to the discovery of important proto-oncogenes and tumour-suppressor genes. We next compiled a list of genes with recurrent somatic mutations. A total of 996 non-silent mutations (see online supplementary tables S2 and S3 for complete lists of point mutations and small indels, respectively) covering 856 genes (see online supplementary table S4) were identified in the 22 patients with CRC. By this ‘recurrent gene’ approach, 52 genes were found to harbour somatic mutations in two or more patients in the discovery cohort. Among these 52 recurrently mutated genes, 5 of them (ie, APC, TP53, KRAS, NF1, FBXW7) have been recorded as cancer genes in Cancer Gene Census (database downloaded on 15 March 2012). We successfully affirmed APC as one of the most frequently mutated genes in our colon cancer series, where non-silent mutation could be detected in 18 out of 22 patients. Non-silent mutations of two other well-known colon cancer-related genes, that is, TP53 and KRAS, were also detected in 9 and 6 out of 22 patients, respectively. In addition, another well-reported CRC-related gene FBXW7 was found to harbour six somatic mutations, namely two missense mutations, two truncations and two frameshift insertions/deletions. However, no mutation was detected in the known CRC driver PIK3CA and the newly discovered, hypermutation-related gene POLE. Gene ontology analysis with Database for Annotation, Visualisation and Integrated Discovery using the data set of 856 genes with non-silent mutations from 22 patients showed significant enrichment of two classical CRC-related signalling pathways, namely ErbB signalling and cadherin/Wnt signalling (p<0.01, false discovery rate (FDR) <5%).
Capture sequencing of 187 genes in a validation cohort of 160 CRC cases
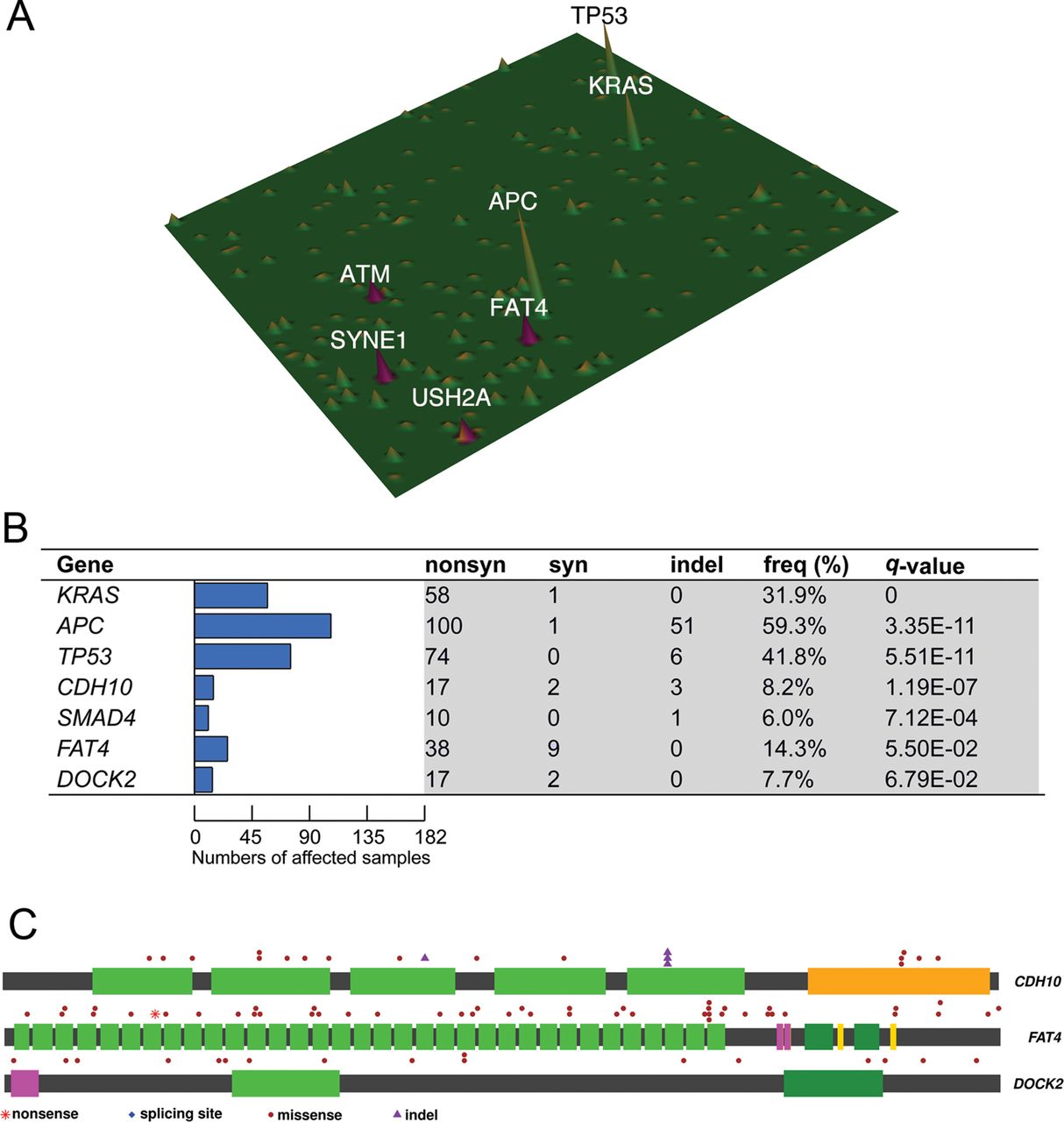
To establish mutation prevalence and clinical relevance of newly identified CRC-related genes, we sequenced the exomic regions of 187 recurrently mutated or pathway-related genes in tumour and blood lymphocytes in an independent cohort of 160 patients with CRC with detailed clinicopathological information by targeted capture sequencing (see online supplementary table S5 for patient information). Target regions were sequenced at a median depth of 126-fold for 160 pairs of CRC genomes and matched lymphocyte controls (see online supplementary figure S2). A bioinformatic approach similar to that of exome sequencing was then conducted to catalogue somatic mutations of these selected genes in the validation cohort. The mutation landscape of 187 captured genes in 160 patients was depicted in figure 3A. Among 160 patients with CRC, 140 cases had at least one non-silent mutation detected in the captured gene set. The number of non-silent somatic mutations in the targeted capture regions among 160 patients with CRC ranged from 0 to 432 with a median of 5 (see online supplementary table S6). Using somatic mutation rate >12 per Mb as a boundary,8 15 patients with CRC were regarded as harbouring hypermutated tumours (see online supplementary figure S3). As expected, APC (56.3%), TP53 (41.9%) and KRAS (32.5%) were the three most frequently, non-silently mutated genes among the 160 patients with capture sequenced CRC. Intriguingly, we observed high prevalence (>5%) of non-silent mutations in multiple genes, including SYNE1 (17.5%), FAT4 (14.4%), ATM (10.6%), USH2A (10.0%), CDH10 (8.8%) and MLL3 (8.8%) (see online supplementary tables S7–9).

Identification of novel high-frequency and significantly mutated genes by targeted capture sequencing in CRC. (A) Mutation landscape of 160 capture-sequenced patients with CRC was depicted in which several novel mutated genes (ie, SYNE1, FAT4, ATM, USH2A) were shown to exhibit mutation frequency of ≥10%. (B) Significantly mutated genes (SMGs) in which non-silent mutations were positively selected over silent mutations were identified in 182 exome-sequenced and capture-sequenced patients with CRC and ranked by q-value. Such analysis reaffirmed APC, KRAS, TP53 and SMAD4 mutations as major driver events in CRC. Our analysis also revealed three novel SMGs, namely FAT4, CDH10 and DOCK2, previously undescribed in CRC. (C) Distribution of somatic mutations in the three newly identified SMGs was shown.
Significantly mutated genes and pathways in CRC
To identify mutated genes that are causally related to and thus positively selected in tumorigenesis, we combined data from exome and capture sequencing to compile a list of SMGs that exhibit higher-than-expected variant counts due to selective advantages by MutSigCV. Such analysis revealed 7 SMGs, namely APC (59.3%), KRAS (31.9%), TP53 (41.8%), FAT4 (14.3%), CDH10 (8.2%), DOCK2 (7.7%) and SMAD4 (6.0%) in 182 patients with CRC (figure 3B; q-value <0.1). Four of these genes, namely APC, TP53, KRAS and SMAD4, have been reported in previous CRC genomic studies whereas the remaining three genes (CDH10, FAT4 and DOCK2) are novel CRC-related genes (figure 3C). The number of potentially protein function-changing mutations identified by SIFT and PolyPhen2 of these seven SMGs was shown in online supplementary table S10. Using Oncodrive, another method for discovering SMGs by assessing functional impacts of mutations,24 additional genes, such as ACTC1, SMAD3 and PIK3R3 were identified (see online supplementary table S11).
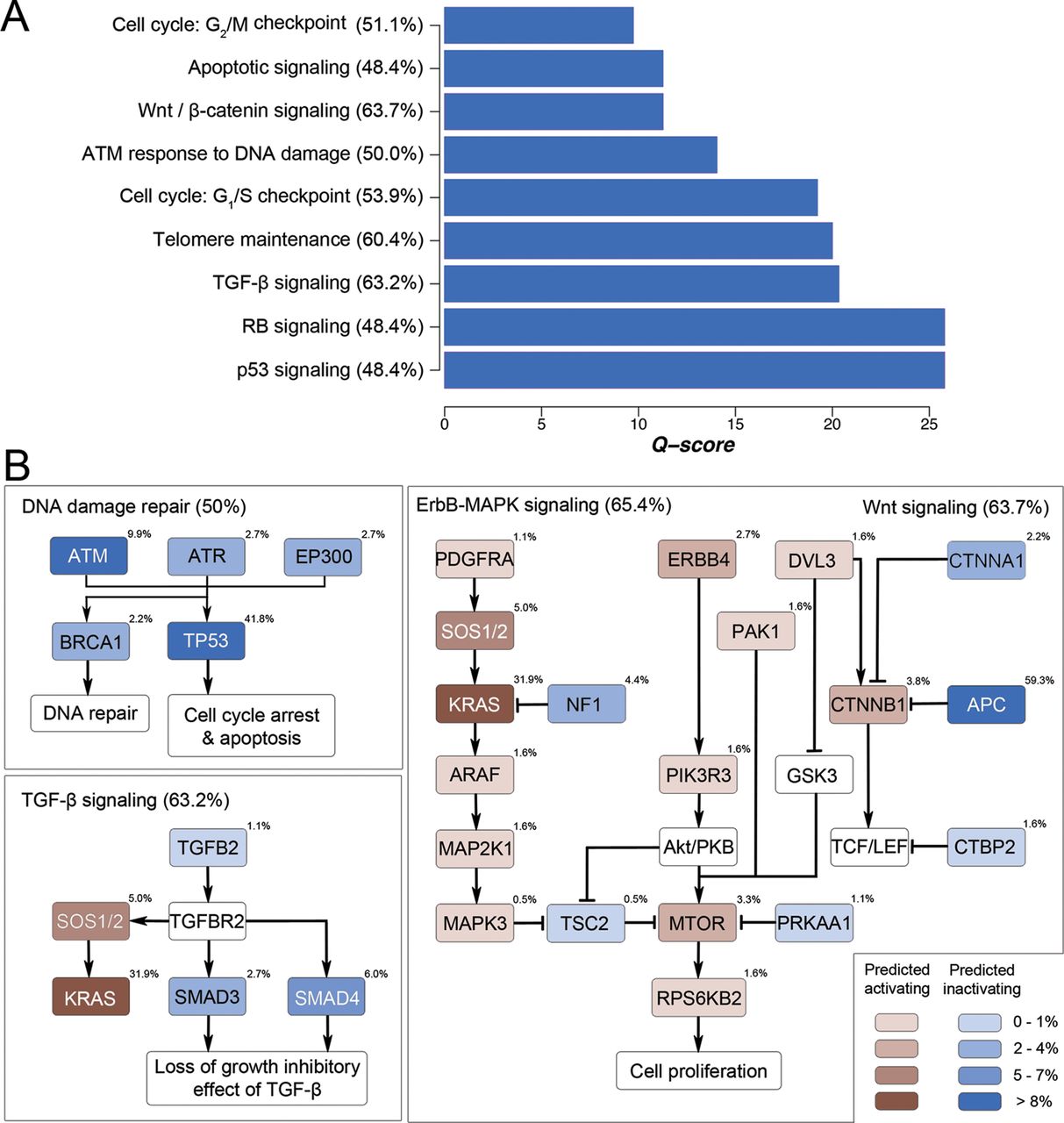
Analysis of mutation frequencies of different signalling pathway components showed that several classical CRC-related pathways were significantly mutated in CRC (figure 4A). Figure 4B illustrates the mutation frequencies of major signalling components in Wnt/β-catenin signalling, ErbB signalling, TGF-β signalling and DNA damage sensing and repair. Concordant with previous findings,25 APC (59.3%) and CTNNB1 (3.8%) mutations accounted for the major genetic abnormalities in the Wnt/β-catenin signalling. A major proportion of patients with CRC (53.9%) also harboured mutations in one or more components in the DNA damage sensing and repair system, including TP53 (41.8%), ATM (9.9%)/ATR (2.7%) (encoding DNA damage-sensing proteins), EP300 (2.7%) (encoding a p53 coactivator) and BRCA1 (2.2%) (encoding a DNA double-strand break repair enzyme). In the ErbB cascade, aside from the preponderant KRAS mutation (31.9%), we observed novel recurrent mutations of NF1 (4.4%), which encodes a putative tumour suppressor protein known as neurofibromin that accelerates guanosine triphosphate hydrolysis and thus inactivation of Ras.26

Signalling pathways genetically altered in CRC. (A) Significantly mutated pathways with positive selection of non-silent mutations were ranked by Q-score. (B) Mutation frequencies of individual signalling components of four major signalling pathways, namely, Wnt signalling, ErbB signalling, transforming growth factor-β signalling and DNA damage sensing/repair, in 182 patients with CRC were shown. These pathways exhibited genetic alteration in a majority (50–65%) of CRC samples.
A five-gene signature for TNM-staging-independent prognostication
To develop a mutation signature marker for prognostication in clinical settings, we constructed a gene signature through combining mutated genes that were associated with better overall survival. Only genes with mutation prevalence ≥5% were included to allow sufficient representation of patients with CRC in signature-positive and signature-negative groups. Using this approach, we developed a signature comprising CDH10, COL6A3, SMAD4, TMEM132D, VCAN in which mutation(s) in one or more of its composing genes occurred in approximately a quarter of patients with CRC. Mutation(s) in this five-gene signature significantly predicted a better overall survival in patients with CRC independent of tumour differentiation and TNM staging in multivariate analysis (table 1). The median overall survival of patients with mutation(s) in this gene signature was 80.4 months in the mutant group versus 42.4 months in the wild type group (p=0.0051; figure 5A). Subgroup analyses in Stage I+II patients revealed that this prognostic mutation signature could be used to stratify patients with CRC with different survival outcomes in early-stage CRC (p=0.0362; figure 5B). Moreover, the mutation prevalence and prognostic significance of this five-gene signature in MSS and MSI-low/high patients with CRC were similar (see online supplementary figure S4A, B). Although KRAS mutations by itself did not have prognostic significance in our cohort, the use of our five-gene signature for predicting survival seems to be more effective in patients with CRC with wild type KRAS genotype than those with mutated KRAS (see online supplementary figure S4C, D). We next verified our prognostic marker in an independent cohort by extracting mutation and survival data from TCGA study.8 Concordant with our finding, mutation(s) in one or more genes in this five-gene signature was significantly associated with better survival in TCGA cohort (p=0.0345; figure 5C). Importantly, such association could be readily observed in patients with early-stage (ie, Stage I+II) CRC (p=0.0106; figure 5D). Multivariate analysis revealed that mutation(s) in the five-gene signature was significantly associated with better overall survival in patients with CRC of TCGA cohort independent of TNM staging and MSI status (see online supplementary table S12). Further subgroup analysis indicated that, after exclusion of MSI cases from TCGA cohort, a consistent association could still be observed (p=0.0258; see online supplementary figure S4E). By combining MSS patients from both cohorts, mutation(s) in the five-gene signature were significantly associated with better survival, indicating that the survival advantage of signature-mutant patients was not secondary to MSI (p=0.0057; figure 5E).
Univariate and multivariate Cox regression analyses of potential correlations between different clinicopathological parameters and survival of patients with CRC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Identification of a five-gene signature (CDH10, COL6A3, SMAD4, TMEM132D, VCAN) that was associated with significantly better overall survival in patients with CRC. Mutation frequency of individual composing genes was ≥5% in our CRC cohort. (A) Kaplan-Meier survival analysis showed that patients with mutation(s) in at least one composing gene of this signature had significantly longer overall survival than those patients with wild type genotype (median survival 80.4 months vs 42.4 months; p=0.005). (B), Subgroup analysis in patients with stage I+II CRC confirmed the prognostic value of this five-gene signature in early stage CRC. (C) The prognostic significance of this five-gene signature was verified in an independent cohort by extracting mutation and survival data from The Cancer Genome Atlas (TCGA) study. (D) The five-gene signature readily differentiated patients with dissimilar survival outcomes in stage I+II CRC in TCGA cohort. (E), The five-gene signature was significantly associated with survival in microsatellite-stable (MSS) patients (Asian+TCGA cohorts), suggesting that survival advantage of signature-mutant patients was not conferred by MSI.
Discussion
Through a two-phase approach for genomic discovery, we aimed at uncovering genes important for CRC pathogenesis. Whole-exome sequencing of 22 CRC genomes followed by large-cohort validation by targeted capture sequencing confirmed APC, KRAS and TP53 mutations as predominant genetic defects in our patient cohort. Our study also identified several previously reported CRC-associated genes (eg, SMAD4, MLL3, CTNNB1, ATM and DCC), which substantiated the importance of these genes in CRC development.8 ,25 ,27 ,28 The mutation spectrum was also consistent with previous studies in which C/G>T/A transition was the most frequently observed nucleotide change.8 Such predominance has been attributed to several factors, including deamination of 5-methylcytosine at CpG islands, deamination of non-methylated cytosines to uracil, and O6-methylation of guanine.29 However, it is worthwhile to notice that the depth of exome sequencing in the first part of our study might be relatively suboptimal and tumour samples were not microdissected and the proportion of ‘contaminating’ normal cells could be high in some cases.
One of the most notable findings of the present study is the identification of novel SMGs that have not been described in CRC. Particularly, one SMG (ie, FAT4) exhibited mutation prevalence of >10% and two SMGs (ie, DOCK2, CDH10) mutated at frequencies of >7%. All three newly identified SMGs (ie, FAT4, CDH10, DOCK2) have been implicated in tumorigenesis. FAT4 is one of the human homologue of Drosophila FAT, which encodes a cadherin-related protein that suppresses tumour formation and activates planar cell polarity signalling (a non-canonical Wnt signalling pathway).30 Epigenetic and genetic mechanisms are involved in the disruption of FAT4 function in human cancers. To this end, promoter hypermethylation of FAT4 has been reported in breast and lung cancers.31 ,32 FAT4 is also recurrently mutated in melanoma and gastric cancer.33 ,34 In the latter, knockdown of FAT4 reduces cell adhesion but strongly induces cell migration and invasion.34 Our study demonstrated for the first time that 14.3% of patients with CRC harbour FAT4 mutation. Similar to FAT4, CDH10 encodes a cadherin protein. Cadherin-10 is a type II classic cadherin that functions in cell-cell adhesion. A previous study has shown that cadherin-10 could bind to β-catenin, a mediator of canonical Wnt signalling.35 Consistent with its putative role as a tumour suppressor, the expression of cadherin-10 is downregulated in prostate cancer in which its expression is extremely low or undetectable.36 In contrast to FAT4 and CDH10, DOCK2 may function as an oncogene. DOCK2 is a guanine nucleotide exchange factor that promotes RAC1 activation. Two recent studies have shown that aberrant RAC1 activation could induce nuclear factor-κB and Wnt/β-catenin signalling in CRC cells.37 ,38 To this end, recurrent mutations of DOCK2 and its partner ELMO1 at multiple loci have been implicated in the abrogation of their autoinhibitory activities, and thereby enhancing RAC1 function, in oesophageal adenocarcinoma.39 These findings hint at the possibility that mutations in FAT4, CDH10 and DOCK2 could destabilise canonical and non-canonical Wnt signalling to promote CRC.
Aside from the identification of novel SMGs and SMPs, we set out to unravel potential correlation between somatic mutation patterns and clinicopathological features so as to devise a clinically applicable prognostic marker. In this respect, we developed a five-gene prognostic mutation marker composed of CDH10, COL6A3, SMAD4, TMEM132D, VCAN. All these genes had a mutation prevalence of ≥5% and about a quarter of patients with CRC in our cohort harboured at least one mutation in these five genes. These patients exhibited excellent prognosis independent of other clinicopathological parameters. Among these genes, CDH10, TMEM132D and VCAN showed mutual exclusivity with KRAS mutations (see online supplementary table S13). These findings suggest that tumours harbouring mutation(s) in this signature might represent a molecular subtype of CRC with distinct prognostic and genetic features. Above all, the prognostic significance of this signature was successfully verified in TCGA cohort. The clinical utilisation of our prognostic marker may help to differentiate patients with CRC with dissimilar survival outcomes and thus more aggressive adjuvant chemotherapy could be given to those with predicted poorer prognosis. Nevertheless, it is noteworthy that heterogeneous treatments could be a caveat in our survival analysis. In clinical settings, the development of a standard kit for targeted capture and the availability of next-generation sequencing instruments are also required for facilitating the application of our prognostic marker.
Taken together, we successfully identified a number of novel SMGs in CRC. Pertinent to clinical practice, a five-gene mutation signature was devised for predicting survival in patients with CRC. These findings represent major breakthroughs in our understanding of the genetic basis of CRC, and have realised the utilisation of genomic data for prognostication.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 10 - Online table S5
- Data supplement 11 - Online table S6
- Data supplement 12 - Online table S7
- Data supplement 13 - Online table S8
- Data supplement 14 - Online table S9
- Data supplement 2 - Online table S1
- Data supplement 3 - Online table S10
- Data supplement 4 - Online table S11
- Data supplement 5 - Online table S12
- Data supplement 6 - Online table S13
- Data supplement 7 - Online table S2
- Data supplement 8 - Online table S3
- Data supplement 9 - Online table S4
Footnotes
-
JY, WKKW and XL are co-first authors and contributed equally.
-
Contributors JY, JJYS, H-FK, HY and JW designed and managed the project. NZ, JC, SSMN, PBSL, JHT, KFT prepared the samples. XL, JH, CY, ZG, JY, ML, QW and YLi performed bioinformatic analysis. X-XL, QL and YP performed the experiments. JY and WKKW analysed the data and wrote the paper. NW, YLu, YLi, FKLC and JJYS revised the paper.
-
Funding The work was supported by Cancer Genome Project of the Chinese University of Hong Kong (2009), Shenzhen Municipal Science and Technology R&D funding (JCYJ JCYJ20120619152326450), China 863 program fund (2012AA02A506), Theme-based Research Scheme of Hong Kong RGC (T12-403-11), China 973 Program fund (2013CB531401) and Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute. The study sponsor did not play any role in the study design and in the collection, analysis and interpretation of data.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The study protocol was approved by the Clinical Research Ethics Committee of the Chinese University of Hong Kong.
-
Provenance and peer review Not commissioned; externally peer reviewed.