Article Text

Abstract
Objective Neurotensin (NT) mediates colonic inflammation through its receptor neurotensin receptor 1 (NTR1). NT stimulates miR-133α expression in colonic epithelial cells. We investigated the role of miR-133α in NT-associated colonic inflammation in vitro and in vivo.
Design miR-133α and aftiphilin (AFTPH) levels were measured by quantitative PCR. Antisense (as)-miR-133α was administrated intracolonicaly prior to induction of 2, 4, 6-trinitrobenzene sulfonic acid (TNBS)-induced colitis and dextran sodium sulfate (DSS)-induced colitis. The effect of AFTPH was examined by gene silencing in vitro.
Results NT increased miR-133α levels in NCM-460 overexpressing NTR1 (NCM460-NTR1) and HCT-116 cells. NT-induced p38, ERK1/2, c-Jun, and NF-κB activation, as well as IL-6, IL-8 and IL-1β messenger RNA (mRNA) expression in NCM-460-NTR1 cells were reduced in miR-133α-silenced cells, while overexpression of miR-133α reversed these effects. MiR-133α levels were increased in TNBS (2 day) and DSS (5 day) colitis, while NTR1 deficient DSS-exposed mice had reduced miR-133α levels, compared to wild-type colitic mice. Intracolonic as-miR-133α attenuated several parameters of colitis as well expression of proinflammatory mediators in the colonic mucosa. In silico search coupled with qPCR identified AFTPH as a downstream target of miR-133α, while NT decreased AFTPH expression in NCM-460-NTR1 colonocytes. Gene silencing of AFTPH enhanced NT-induced proinflammatory responses and AFTPH levels were downregulated in experimental colitis. Levels of miR-133α were significantly upregulated, while AFTPH levels were downregulated in colonic biopsies of patients with ulcerative colitis compared to controls.
Conclusions NT-associated colitis and inflammatory signalling are regulated by miR-133α-AFTPH interactions. Targeting of miR-133α or AFTPH may represent a novel therapeutic approach in inflammatory bowel disease.
- IBD BASIC RESEARCH
- COLONIC DISEASES
- EXPERIMENTAL COLITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Neurotensin (NT) mediates colonic inflammation through its high-affinity receptor (neurotensin receptor 1 (NTR1)).
NT and NTR1 expressions are upregulated in inflamed colon tissues in experimental colitis and ulcerative colitis (UC).
NT modulates differential expression of 38 microRNAs in colonocytes, among them miR-133α.
MicroRNA-133α modulates signalling pathways related to proliferation, differentiation and hypertrophy in cancer cell lines and myocytes.
What are the new findings?
miR-133α modulates NT-induced MAP kinase, and NF-κB activation, as well as transcription of proinflammatory cytokines in vitro.
Reducing the levels of miR-133α in human colonocytes and mouse colon attenuates proinflammatory signalling and development of experimental colitis in vitro and in vivo.
Aftiphilin (AFTPH) is a novel downstream target of NT-induced miR-133α expression in human colonic epithelial cells involved in proinflammatory signalling pathways.
Colon tissues from patients with UC and mice with colitis have higher miR-133α but lower AFTPH levels when compared with controls.
How might it impact on clinical practice in the foreseeable future?
Inhibiting miR-133α and restoring AFTPH expression in inflammatory bowel disease (IBD) colon may represent promising approaches for the treatment of IBD.
The reverse association of the levels of miR-133α and AFTPH together with increased expression of NTR1 in UC may represent novel biomarkers for UC.
Introduction
Neurotensin (NT) is a neuropeptide expressed in the central nervous system and the intestine,1 ,2 while its high-affinity receptor, NT receptor 1 (NTR1)3 is expressed in neurones,4 colonic epithelial,2 ,5–7 immune cells,8 ,9 as well as different colon cancer cell lines.10 In the colon, NT/NTR1 signalling promotes inflammation in acute colitis animal models.2 ,5 Colonic NT and NTR1 expression increases during enterotoxin-mediated intestinal inflammation,2 dextran sodium sulfate (DSS) colitis,6 ,11 ,12 and in the colon of patients with ulcerative colitis (UC).6 Moreover, NTR1 deficiency in mice is associated with reduced weight loss and mortality in experimental colitis.5 .13
MicroRNAs (miRs) are short (19–25 nucleotides), single-stranded RNA molecules, acting as negative transcriptional regulators. They bind to the 3′ untranslated regions (UTR) of transcripts14 and lead to messenger RNA (mRNA) degradation, or inhibition of translation into protein.15 Recently, several studies identified the differential miR expression in colonic mucosa in patients with UC,16–19 Crohn's Disease (CD)18 ,20 and experimental colitis.17 However, the role of individual miRs in inflammatory bowel disease (IBD) pathophysiology is still under investigation.
We have recently shown that NT/NTR1 coupling in human colonic epithelial NCM460 overexpressing NTR1 (NCM460-NTR1) cells induces differential expression of miRs,10 including miR-133α. Dysregulation of miR-133α and its target genes has been correlated with colorectal cancer development21–28 through regulating ERK29 ,30 and Akt21 ,31 activation, pathways also targeted by NT/NTR1 signalling in human colonic epithelial cells.7 ,10 ,32 Based on these considerations, we examined the role of NT-miR-133α interactions in colonic inflammation in vitro and in two experimental colitis models induced by trinitrobenzene sulfonic acid (TNBS) and DSS in vivo. We also performed studies to identify the target of NT-NTR1-modulated miR-133α that mediates these effects. Our results point to aftiphilin (AFTPH), as a novel downstream target for miR-133α, and present evidence for the association of miR-133α and its downstream target, AFTPH, in the pathophysiology of colitis.
Materials and methods
Cell culture and reagents
NCM460-NTR1 cells were generated from human colonic epithelial NCM460 cells (INCELL, San Antonio, Texas, USA) transduced with lentivirus particles as previously described10 and maintained in M3:D culture media (INCELL) supplemented with 10% fetal bovine serum (FBS, Life Technologies, Grand Island, New York, USA). Human embryonic kidney fibroblasts, HEK293, and colonic cancer HCT-116 cells were maintained in Eagle's Minimum Essential Medium (MEM, Life Technologies) and McCoy5a (ATCC, Manassas, Virginia, USA), respectively, and supplemented with 10% FBS. Lipofectamine 2000, lipofectamine RNAimax and OptiMEM were from Life Technologies. NT was from Bachem Americas (Torrance, California, USA). TNBS and SR48962 were from Sigma Aldrich (St Louis, Missouri, USA), and DSS was from MP Biomedicals (Santa Ana, California, USA). Goat anti-AFTPH (sc-167055), mouse antivillin and rabbit anti-β tubulin were from Santa Cruz Biotechnology (Santa Cruz, California, USA).
Transfection experiments
Small interfering RNA (siRNA) was purchased from Santa Cruz Biotechnology (Santa Cruz). NCM460-NTR1cells were transfected with siRNA against AFTPH (si-AFTPH) using Lipofectamine RNAiMAX. All miRs were purchased from Life Technologies. For miR-133α silencing or overexpression, cells were transfected with antisense-miR-133α (as-miR-133α) or miR-133α precursor (miR-133α), respectively, using Lipofectamine RNAiMAX. Cells transfected with siRNA-A (si-control), antisense-control miR (as-miR-control) or miR-negative control precursor (miR-control) served as controls.
NT/NTR1-mediated phosphoprotein activation and cytokine production
NCM460-NTR1 cells were transfected with as-miR-133α, miR-133α and si-AFTPH and their respective controls. Cells were incubated in serum-free media overnight and stimulated with NT (100 nM). Cell lysates were collected for phosphoprotein detection, and media were collected 6 h after stimulation for cytokine neasurements. Custom designed Bio-Plex Pro Cell Signalling assay panel (Bio-Rad, Hercules, Californai, USA) and Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad) were used for phosphoprotein activation and cytokine production (except IL-8), respectively, according to manufacturer's instructions.
Animal models and establishment of experimental colitis models
Neurotensin receptor 1 knockout mouse model
Ntsr1tmDgen (hereafter called NTR1KO) were purchased from Jackson Laboratories and bred in our facility. Animals were obtained as fifth generation backcross of 129 onto C57BL6/J. We performed one additional backcross before intercrossing animals as heterozygous NTR1KO crosses to generate littermate controls.
TNBS-induced colitis
C57BL6/J wild-type (WT) mice were purchased from Jackson Laboratories. TNBS was dissolved in 30% ethanol (5 mg/kg) was intracolonicaly administered (50 μL) to mice as previously described.5 ,33 Briefly, TNBS was slowly infused via a 1 mL syringe (Becton Dickinson, Laguna Hills, California, USA) fitted with a polyethylene cannula (Intramedic PE-20 tubing; Becton Dickinson). Colon tissues were collected 48 h after TNBS administration.
DSS-induced colitis
NTR1KO and its WT counterparts, or C57BL6/J WT animals, were treated with 5% DSS in drinking water as previously described,13 ,34 and colon tissues were collected 5 days after the start of the treatment.
In vivo NT infusion. NT was dissolved in PBS supplemented with 1% bovine serum albumin (BSA)35 and was administered intracolonicaly (300 μg/kg) to C57BL6/J WT mice twice per day for 4 days. Control mice were infused with 1% BSA in PBS. Colon tissues were collected 5 days after NT administration.
In vivo knockdown of miR-133α
Endogenous expression of miR-133α in colon tissues of C57BL6/J WT mice was silenced by intracolonic administration of miRCURY LNA Inhibitor probe, in vivo against mmu-miR-133a (20 μg/mouse, Exiqon). Briefly, the appropriate amount of oligonucleotides against mmu-miR-133a and its respective control were resuspended in 100 μL Opti-MEM with 2 μL lipofectamine 2000 and administered intracolonicaly 24 h and 72 h prior to TNBS or DSS treatment.
MicroRNA in situ hybridisation
Colon tissues (3 cm from distal end) were obtained from WT mice 2 day after TNBS treatment. The tissues were immediately fixed with 4% paraformaldehyde, paraffin-embedded and sectioned (5 μm) for histological study. 5′-DIG and 3′-DIG labelled detection probes specific for mouse miR-133α (Cat. No. 39270-15) and miRCURY LNA microRNA ISH optimisation Kit (FFPE) were purchased from Exiqon, and the experiments were performed according to the manufacturer's instructions.
RNA expression studies in patient samples
Total RNAs from colon tissues obtained from patients with UC (n=12), CD (n=8) and normal subjects (n=9) were purchased from Origene (Rockville, Maryland, USA). Conversion of cDNA of RNA samples was performed as described above, and levels of miR-133α and AFTPH were determined by qPCR analysis.
Immunohistochemistry
Frozen tissue sections (5 μm) obtained from chronic active patients with UC (n=3) and their normal control (n=3) were purchased from Origene. The tissues were fixed with 4% paraformaldehyde and blocked with 0.3% Triton X-100 in PBS (PBS-Triton) supplemented with 5% normal bovine serum. Appropriate antibodies were incubated with the tissue sections overnight at 4°C, washed with PBS-Triton and incubated with appropriate secondary antibodies (Santa Cruz Biotechnology). The tissues were imaged using Axio Imager.Z1 microscope (Carl Zeiss).
Please refer to online supplementary methods for site-directed mutagenesis, luciferase assay, NF-κB p65 translocation, measurement of interleukin-8 (IL-8) production, immunoblot analysis, mRNA and miR expression analysis and microRNA profiling in blood samples.
Statistical analysis
All in vitro results derived from at least three sets of experiments, expressed as means±SD and analysed with Student t tests. Results from animal experiments and studies in human tissues were analysed with Student t tests and expressed as means±SEM. In all statistical comparisons, p<0.05 was used to indicate significant differences.
Results
Increased miR-133α levels in human colonic epithelial cells after NT exposure
NT exerts its effect on colonic epithelial cells2 ,5–7 through its high-affinity receptor NTR1.3 NTR1 overexpression is observed in colon cancer cell lines,10 ,36 colon tissues from mice with experimental colitis,6 ,11 ,12 intestinal tumors37 and in tissue biopsies from patients with UC.6 In a microarray analysis, we previously showed that NT/NTR1 coupling in human colonic epithelial NCM460-NTR1 increased miR-133α expression.10 In our current study, we first verified this result by examining the levels of miR-133α in human colonic epithelial NCM460-NTR1 cells and in colon cancer adenocarcinoma HCT-116 cells by qPCR. Consistent with our previous findings,10 levels of miR-133α were upregulated after NT exposure by 2.6±0.43-fold (p<0.01, figure 1A) in NCM460-NTR1 cells, while in HCT-116 cells, levels of miR-133α were increased by 1.5±0.07-fold (p<0.01, figure 1B) after addition of NT. Therefore, NT increases miR-133α expression in two different human colonic epithelial cell lines.

Levels of miR-133α were upregulated after neurotensin (NT) exposure in human colonic epithelial cells. (A) Expression of miR-133α in human colonic epithelial NCM460 cells after NT exposure was analysed by qPCR analysis. **p<0.01, compared to vehicle control. (B) miR-133α levels in human colon cancer HCT-116 cells after NT exposure were analysed by qPCR analysis. **p<0.01, compared to vehicle control.
NT induces proinflammatory signalling in human colonic epithelial cells via miR-133α expression
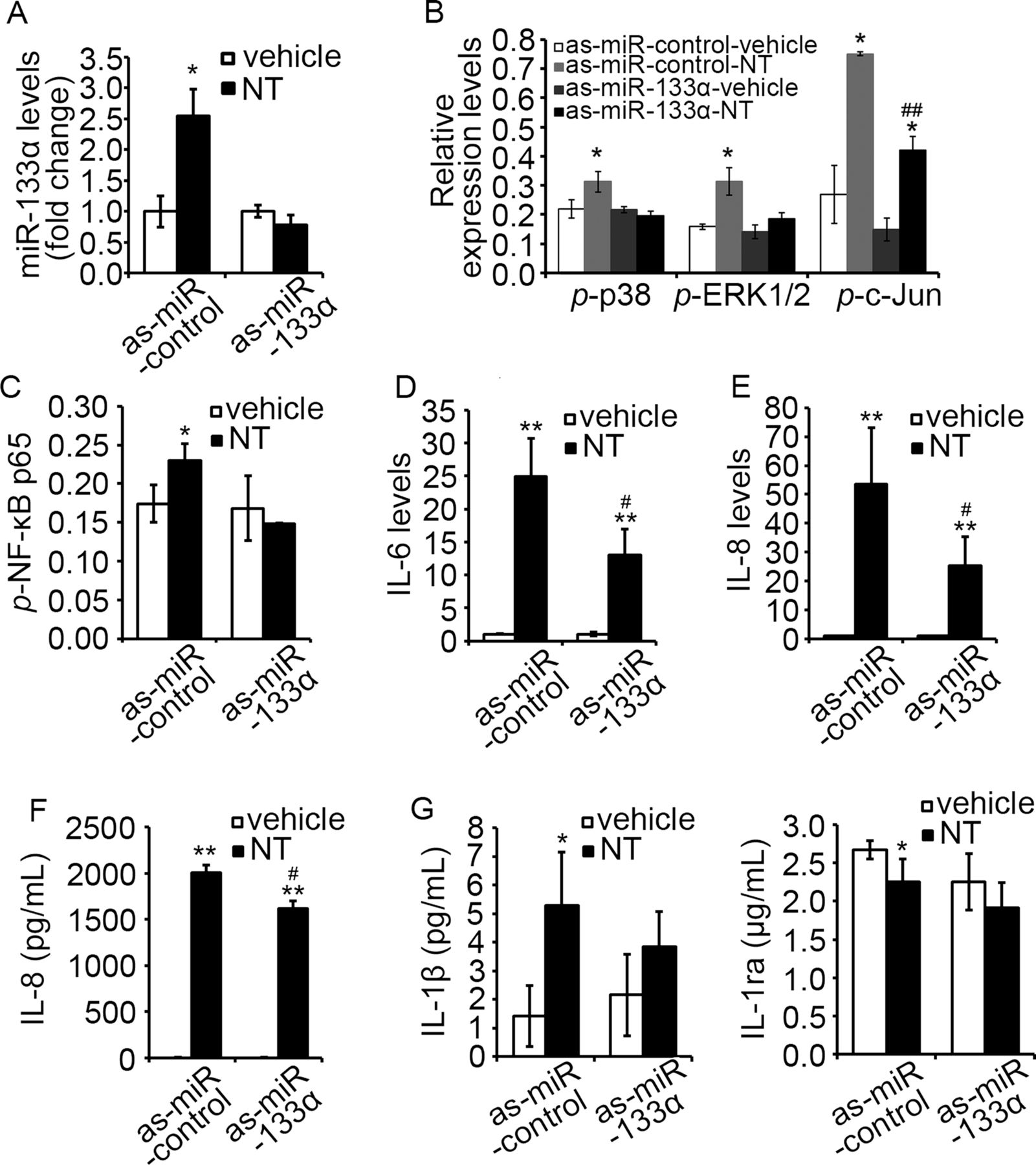
NT coupling of NTR1 stimulates inflammatory responses through MAPK38 ,39-dependent and NF-κB7 ,40 ,41-dependent pathways. Here, we examined whether miR-133α is involved in NT-related proinflammatory signalling and cytokine expression in NCM460-NTR1 cells with an antisense approach using as-miR-133α. First, we showed that transfection of as-miR-133α blocked NT-induced miR-133α overexpression in NCM460-NTR1 cells (figure 2A), compared to those transfected with control as-miR, validating this approach. Moreover, NT stimulation activated p38 and ERK1/2 signalling only in cells transfected with control as-miR, but not as-miR-133α, while NT-induced c-Jun activation was significantly attenuated in as-miR-133α-transfected cells (figure 2B, p<0.05). Moreover, NT-induced NF-κB p65 phosphorylation was blocked in as-miR-133α-transfected cells (figure 2C), while NT-induced IL-6 and IL-8 mRNA (figure 2D, E, p<0.05) and IL-8 secretion were also significantly attenuated upon miR-133α knock-down (figure 2F, p<0.05). Analysis of the cell culture media showed that NT stimulation increased the production of the proinflammatory IL-1β, but reduced the production of the anti-inflammatory IL-1ra (figure 2G, p<0.05). Transfection of as-miR-133α significantly reduced NT-induced IL-1β production, while prevented the decrease in IL-1ra production (figure 2G). Together, these results suggest an important role for miR-133α in the induction of proinflammatory signalling pathways and cytokine production in response to NTR1 activation in human colonocytes.

Downregulation of miR-133α attenuated neurotensin (NT)-associated proinflammatory signalling in NCM460-neurotensin receptor 1 (NTR1) cells. (A) Levels of miR-133α in NCM460-NTR1 cells upon NT stimulation after transfection of as-miR-133α and its control were analysed by qPCR analysis. *p<0.05, compared to cells transfected with as-miR-control. (B) Levels of MAP kinase phosphorylation, including p38, ERK1/2 and c-Jun and (C) NF-κB phosphorylation in NCM460-NTR1 cells transfected with as-miR-133α and its control were analysed as stated in Methods. *p<0.05, compared to cells transfected with as-miR-control; ##p<0.01, compared to cells treated with NT. (D) IL-6 and (E) IL-8 levels in NCM460-NTR1 cells transfected with as-miR-133α and its control was analysed by qPCR analysis. **p<0.01, compared to cells transfected with as-miR-control and #p<0.05, compared with NT-treated cells. (F) IL-8 production in NCM460-NTR1 cells transfected with as-miR-133α and its control was measured using ELISA. **p<0.01, compared to cells treated with vehicle control; #p<0.05, compared with NT-treated cells. (G) IL-1β and IL-1ra production in NCM460-NTR1 cells transfected with as-miR-133α and its control was measured as stated in Methods. *p<0.05, compared to cells treated with vehicle control.
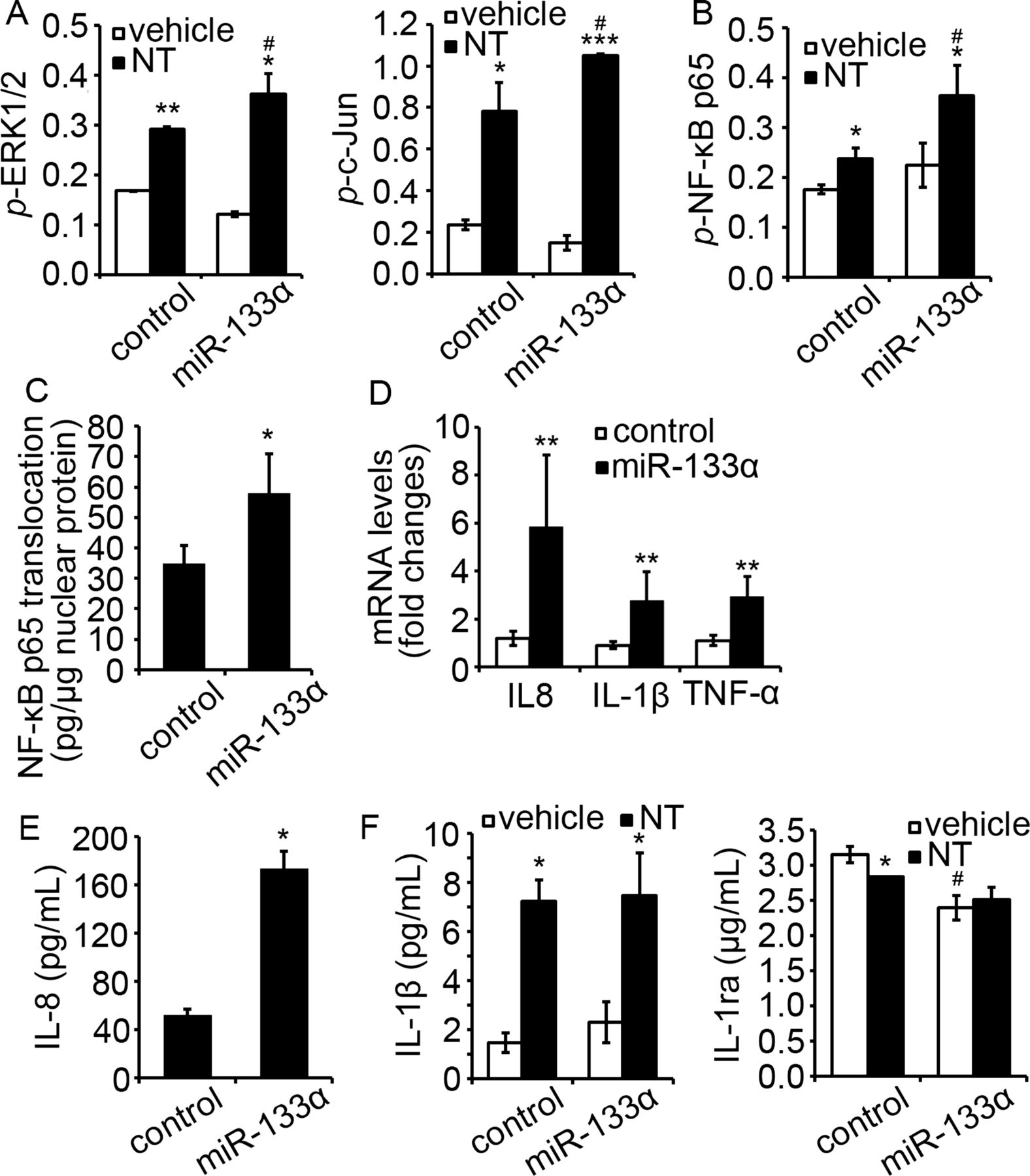
We further verified the proinflammatory role of NT and miR-133α in NCM460 cells transfected with NTR1 and miR-133α. Overexpression of miR-133α further increased NT-induced ERK1/2, c-Jun (figure 3A, p<0.05) and NF-κB p65 (figure 3B, p<0.05) activation. Additionally, overexpression of miR-133α alone, in the absence of NT stimulation, promoted nuclear translocation of NF-κB p65 (figure 3C, p<0.05) when compared to the cells expressing control miR. Furthermore, IL-8, IL-1β and TNF-α mRNA was increased in cells overexpressing miR-133α when compared to their controls (figure 3D, p<0.05). For cytokine production, IL-8 secretion was increased upon miR-133α overexpression (figure 3E, p<0.05). Cells overexpressing miR-133α had also significantly reduced IL-1ra production (p<0.05), while IL-1β production was increased slightly, but not significantly (figure 3F). Together, these results strongly suggest an important role for miR-133α in proinflammatory signalling in human colonocytes.
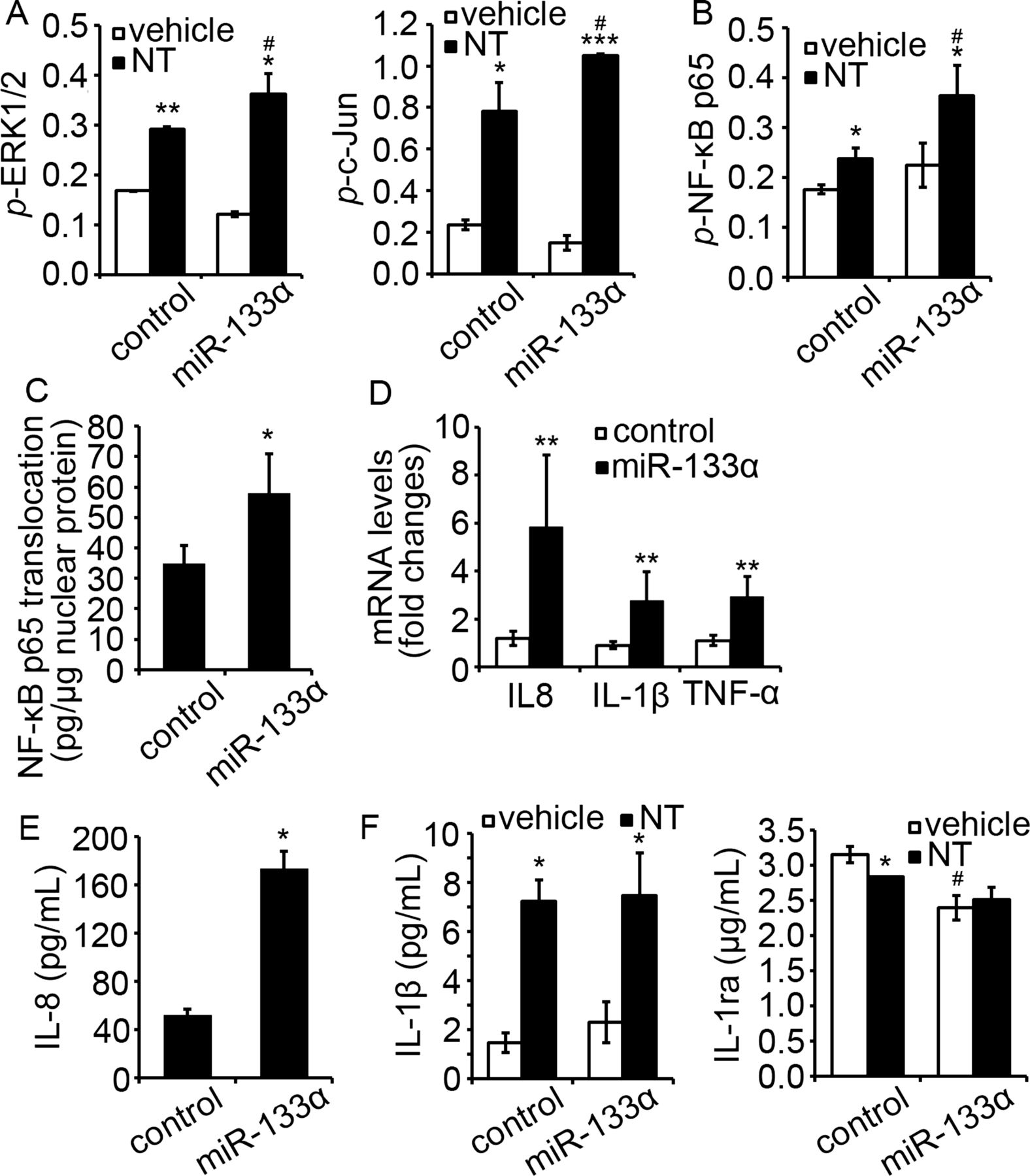
Overexpression of miR-133α promoted proinflammatory signaling in NCM460-neurotensin receptor 1 (NTR1) cells. (A) Levels of MAP kinase phosphorylation, including ERK1/2 and c-Jun and (B) NF-κB p65 phosphorylation in NCM460-NTR1 cells transfected with miR-133α and its control was analysed as stated in Methods. *p<0.05, compared to cells transfected with control miRNA precursor. (C) Translocation of NF-κB p65 in NCM460-NTR1 cells transfected with miR-133α and its control was analysed by ELISA. *p<0.05, compared to cells transfected with control miRNA precursor. (D) Expression of IL-8, IL-1β and TNF-α in NCM460-NTR1 cells transfected with miR-133α and its control was analysed by qPCR analysis. *p<0.05, compared to cells transfected with control miRNA precursor. (E) IL-8 production in NCM460-NTR1 cells transfected with miR-133α and its control was measured using IL-8 ELISA. *p<0.05, compared to cells transfected with control miRNA precursor. (F) IL-1β and IL-1ra production in NCM460-NTR1 cells transfected with miR-133α and its control was measured as stated in Methods. *p<0.05, compared to cells treated with vehicle control; #p<0.05, compared to cells transfected with miR control precursor.
MiR-133α overexpression in vivo is NT/NTR1-dependent
Since, as shown above, NT regulates miR-133α in colonocytes in vitro, we next examined whether this effect can be recapitulated in vivo. To do this, C57/BL6J WT (WT) mice received intracolonicaly NT (300 μg/kg).35 After 4 days, mice were euthanased and colonic tissues were processed for RNA purification. qPCR analysis showed increased colonic miR-133α levels (by 1.3±0.04-fold, figure 4A, p<0.05) in mice administered with NT, compared to intracolonic administration of vehicle. Because NT is an important mediator of acute colonic inflammation,2 ,13 we also examined colonic levels of expression of miR-133α in two colitis models. Acute colitis was induced in C57BL6/J WT mice by intracolonic administration of 5 mg/kg TNBS, or by addition of 5% DSS in their drinking water. qPCR analysis showed increased miR-133α levels (by 3.2±1.01-fold, figure 4B, p<0.05) 2 days after TNBS administration, but decreased by 63% at day 7 (figure 4B, p<0.05). Mice receiving DSS showed a 3.6±0.61-fold increase in miR-133α levels (figure 4B, p<0.001) at the end of the DSS treatment, compared to controls.

Levels of miR-133α were upregulated during experimental colitis in vivo. (A) miR-133α levels in colon tissues from C57BL6/J mice intracolonically administered with NT (300 μg/kg) was analysed by qPCR analysis. *p<0.05, compared to mice receiving vehicle control. (B) Expression of miR-133α in colon tissues from C57BL6/J mice collected 2 and 7 days after 5 mg/kg trinitrobenzene sulfonic acid (TNBS) administration and 5 days after 5% dextran sodium sulfate (DSS) feeding was analysed by qPCR analysis. *p<0.05, compared to treatment control. (C) Representative images of in situ hybridisation of miR-133α of colon tissues from TNBS-treated C57BL6/J mice and their control counterparts (arrows, epithelial cells; arrow heads, lamina propria cells infiltration). Scale: 50 μm. (D) Expression of miR-133α in colon tissues from neurotensin receptor 1 (NTR1) KO and its control counterparts collected 5 days after 5% DSS feeding was analysed by qPCR analysis.*p<0.05, compared to wild-type mice with same treatment.
To study the mucosal distribution of miR-133α in colon tissue during colitis, in situ hybridisation against miR-133α in colon tissue sections obtained from TNBS-treated WT mice and their control counterparts was performed as described in Methods. Our result show that while miR-133α expression was higher in inflamed colons, the majority of miR-133α expression was localised in epithelial cells (figure 4C, arrows) and less in other cells of the lamia propria (figure 4C, arrowheads).
The direct relationship of endogenous NT/NTR1 signalling on miR-133α expression in colon tissues during colitis development was further investigated in NTR1-deficient NTR1KO mice. NTR1KO mice and their WT counterparts were allowed access to 5% DSS in their drinking water, and after 5 days the mice were euthanased and miR-133α levels were measured. As shown in figure 4D, colonic miR-133α levels were increased in WT mice after DSS treatment (2.4±0.32-fold, p<0.01), but decreased in NTR1KO mice (p<0.05). Thus, increased miR-133α expression during acute colitis involves NT/NTR1 interactions.
Intracolonic silencing of miR-133α attenuates the development of acute colitis
We next determined the role of miR-133α in the development of experimental colitis. C57BL6/J WT mice were administered intracolonicaly two doses of as-miR-133α or its control 24 h and 72 h prior to intracolonic administration of TNBS (5 mg/kg) or vehicle. Colonic tissues were harvested 48 h post-TNBS-treatment for gene expression and histological analysis. Histological examination on colon tissues collected from mice without TNBS-treatment did not show any differences (figure 5A). As expected, TNBS administration induced acute colonic inflammation in mice administered with as-miR-control, while administration of as-miR-133α prior to TNBS treatment attenuated colitis development (figure 5A). Additionally, quantitative PCR analysis showed that intracolonic administration of as-miR-133α significantly reduced endogenous colonic expression of miR-133α (p<0.05, figure 5B). Mice treated with as-miR-133α and TNBS showed a significant reduction in colon weight after normalisation against colon length (figure 5C, p<0.05), neutrophil infiltration, and an improvement in mucosal integrity which resulted in a significantly improved histological score (figure 5D, p<0.05). Proinflammatory cytokine production was generally increased in colon tissues treated with TNBS. Pretreatment with as-miR-133α prior to TNBS administration blocked increased expression of the neutrophil product lipocalin 2 (lcn2), and TNF-α, a major proinflammatory cytokine in colitis. The production of cxcl1, an important neutrophil chemoattractant (figure 5E, p<0.05) was significantly reduced in mice administered with as- miR-133α, when compared to control as-miR-administered counterparts (figure 5E). We also investigated the effect of as-miR-133α treatment in the DSS-induced colitis model. Administration of as-miR-133α and its control was performed at 24 h and 72 h before DSS treatment. Mice administered with as-miR-133α and DSS showed reduced weight loss 5 days after DSS treatment (see online supplementary figure S1A, p<0.05) while levels of TNF-α and IL-1β were increased 5 days after DSS-induced colitis and significantly reduced in mice administered with as-miR-133α (see online supplementary figure S1B, C, p<0.05). In conclusion, these results suggested that miR-133α is involved in the pathophysiology of colitis.

Intracolonic administration of as-miR-133α attenuated trinitrobenzene sulfonic acid (TNBS)-induced colitis development in wild-type mice. (A) Representative images from H&E staining of colon tissues from C57BL6/J mice intracolonically administered with as-miR-133α and its control with or without TNBS treatment. Scale: 50 μm. (B) Levels of miR-133α in colon tissues from C57BL6/J mice intracolonically administered with as-miR-133α and its control were analysed using qPCR analysis. *p<0.05, compared to mice administered with control as-miRNA. (C) Colon weight from C57BL6/J mice intracolonically administered with as-miR-133α and its control with or without TNBS treatment were measured and normalised by colon length. *p<0.05, compared to mice administered with control as-miRNA. (D) Total histological score, neutrophil infiltration and mucosal integrity of colon tissues from C57BL6/J mice intracolonically administered with as-miR-133α and its control with TNBS treatment were scored. *p<0.05, **p<0.01, ***p<0.005 compared to mice administered with control as-miRNA. (E) Levels of lcn2, TNF-α and cxcl1 in colon tissues from C57BL6/J mice intracolonically administered with as-miR-133α and its control with or without TNBS treatment were analysed using quantitative PCR analysis. *p<0.05, compared to mice administered with control as-miRNA.
MiR-133α directly regulates AFTPH expression through binding its 3′ UTR in colonic epithelial cells
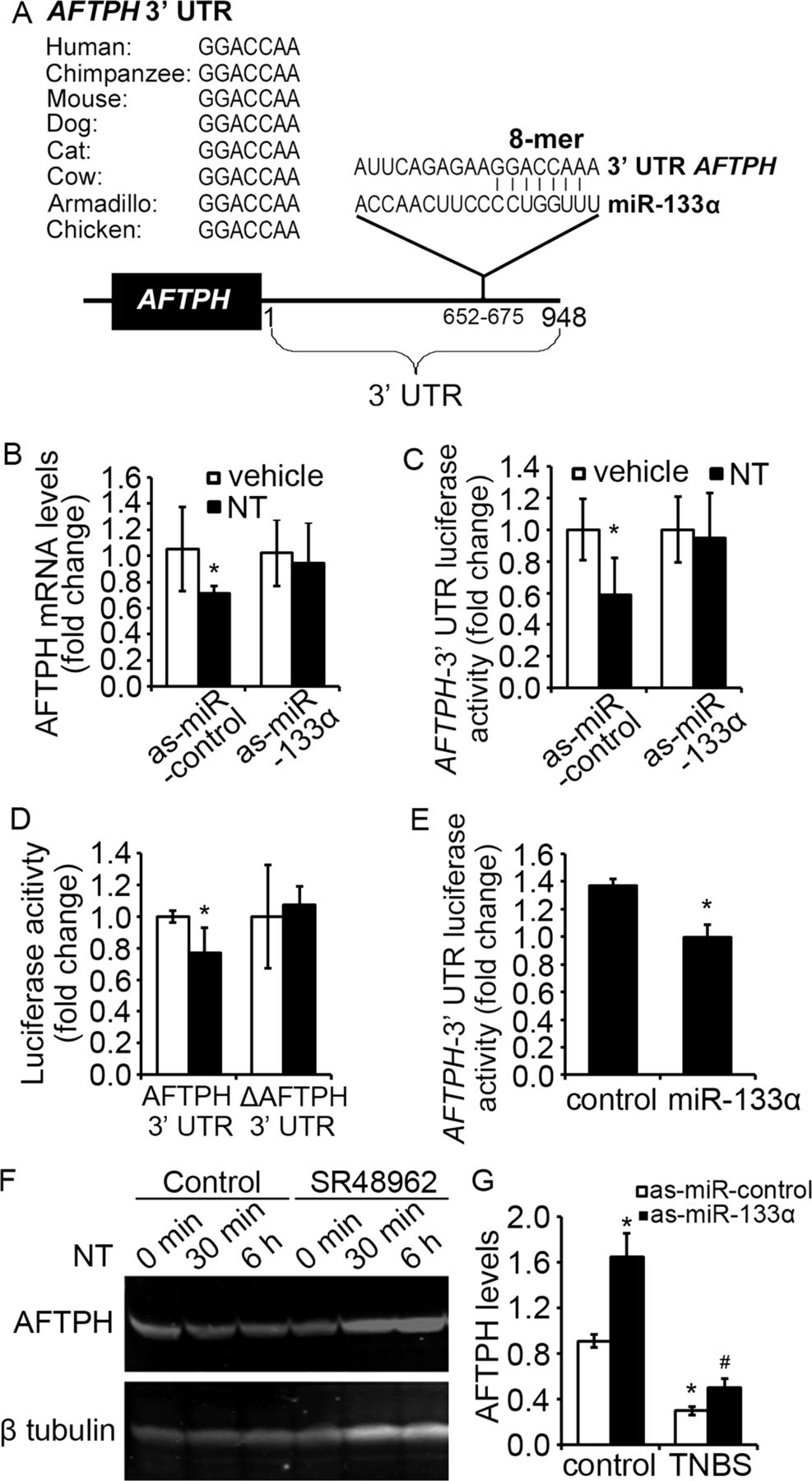
MiRs act as gene-silencers by inhibiting the expression of mRNA transcripts through binding to the 3′ UTRs of target genes.14 To identify candidate genes involved in the NTR1-miR-133α response, we searched for genes with possible miR-133α binding sites in their 3′ UTRs. In silico search using three online databases (TargetScanHuman (http://www.targetscan.org); miRBase (http://www.mirbase.org) and PicTar (http://pictar.mdc-berlin.de)), identified aftiphilin (AFTPH) with a miR-133α binding site at its 3′ UTR highly conserved across species (figure 6A).

Aftiphilin (AFTPH) was the downstream target of neurotensin (NT)-induced miR-133α upregulation. (A) Conservative miR-133α binding sites were identified in 3′ untranslated region (UTR) region of aftiphilin across different species. (B) Levels of miR-133α in NCM460-neurotensin receptor 1 (NTR1) cells transfected with as-miR-133α and its control was analysed by qPCR analysis. *p<0.05, compared to cells stimulated by vehicle. (C) AFTPH 3′UTR-driven luciferase activities from NCM460-NTR1 cells transfected with as-miR-133α and its control were measured after NT stimulation. *p<0.05, compared to cells stimulated by vehicle. (D) Luciferase activity driven by AFTPH 3′UTR with or without miR-133α binding sequence was measured after NT stimulation. *p<0.05, compared to cells stimulated by vehicle. (E) AFTPH 3′UTR-driven luciferase activities from HEK293 cells transfected with miR-133α precursor (miR-133α) and its control were measured. *p<0.05, compared to cells transfected with control miRNA precursor. (F) Immunoblot analysis of AFTPH expression in NCM460-NTR1 cells 30 min and 6 h after NT exposure in the presence or absence of SR48962 (10 nM). (G) AFTPH levels in colon tissues from C57BL6/J mice intracolonically administered with NT and its control and (H) from C57BL6/J mice intracolonically administered with as-miR-133α and its control with or without trinitrobenzene sulfonic acid treatment were analysed using qPCR analysis. *p<0.05, compared to mice administered with control as-miRNA.
The NTR1-miR-133α-AFTPH interaction was first validated in NCM460-NTR1 cells transfected with as-miR-133α and exposed to NT. NT exposure downregulated AFTPH mRNA levels, as shown in quantitative PCR analysis (figure 6B, p<0.05) and AFTPH 3′UTR-regulated luciferase activity assay (figure 6C, p<0.05) when compared with control cells. To confirm the direct interaction between miR-133α and AFTPH, we deleted the miR-133α binding sequence on the AFTPH 3′ UTR and showed that NT-induced downregulation of AFTPH 3′ UTR luciferase activity was not observed in the absence of the miR-133α binding site (figure 6D). Overexpression of miR-133α in HEK293 cells significantly reduced AFTPH-3′ UTR-associated luciferase activity in the absence of NT stimulation (figure 6E, p<0.05). Moreover, treatment with the NTR1-specific antagonist of SR48962 prevented the reduction in AFTPH protein expression induced by NT (figure 6F). The above results strongly indicate that AFTPH is a downstream target of miR-133α in colonocytes after NT exposure, suggesting that NT/NTR1 signalling suppresses AFTPH expression through miR-133α overexpression.
To confirm our in vitro findings in the in vivo setting, we compared expression of AFTPH mRNA in colon tissues from control, NT-exposed and TNBS-exposed (2d) mouse colon. Our results show significantly decreased colonic AFTPH mRNA expression in NT-administered (figure 6G, p<0.05) and TNBS-injected mice (figure 6H, p<0.05) compared to vehicle-exposed control colon, consistent with the increased miR-133α levels observed in both models (figures 4A and 5F). In NTR1KO mice exposed to DSS, only a small (by 1.28±0.17-fold, n=5 per group) but not significant increase in AFTPH levels were observed. Along these lines, silencing of miR-133α by intracolonic administration of as-miR-133α increased AFTPH levels in normal and inflamed colon tissues, when compared to their control counterparts (figure 6H, p<0.05).
AFTPH gene silencing promotes colonic proinflammatory responses in human colonic epithelial cells
To examine the functionality of AFTPH in NT-mediated proinflammatory signalling, we silenced expression of AFTPH in NCM460-NTR1 cells with si-RNA transfection prior to NT stimulation. Our results show that AFTPH gene silencing enhanced NT-induced c-Jun and NF-κB activation (figure 7A, B, p<0.05), consistent with our findings for a role of miR-133α in NT-mediated signalling activation. Additionally, gene silencing of AFTPH increased basal IL-1β production (figure 7C, p<0.05), while ameliorated NT-mediated IL-1ra reduction in human colonic epithelial cells (figure 7C), in line with our results from miR-133α overexpression (figure 3F).

Gene silencing of aftiphilin (AFTPH)-promoted neurotensin (NT)-associated proinflammatory response in vitro. Levels of (A) c-Jun and (B) NF-κB phosphorylation in NCM460-neurotensin receptor 1 (NTR1) cells transfected with si-AFTPH and its control was measured as stated in Methods. *p<0.05, compared to cells transfected with control siRNA; #p<0.05, compared to NT-treated cells. (C) IL-1β and IL-1ra production of NCM460-NTR1 cells transfected with si-AFTPH and its control in the presence or absence of NT stimulation was analysed as stated in Methods. *p<0.05, compared to cells transfected with control siRNA.
Levels of miR-133α and AFTPH are dysregulated in colon of patients with UC
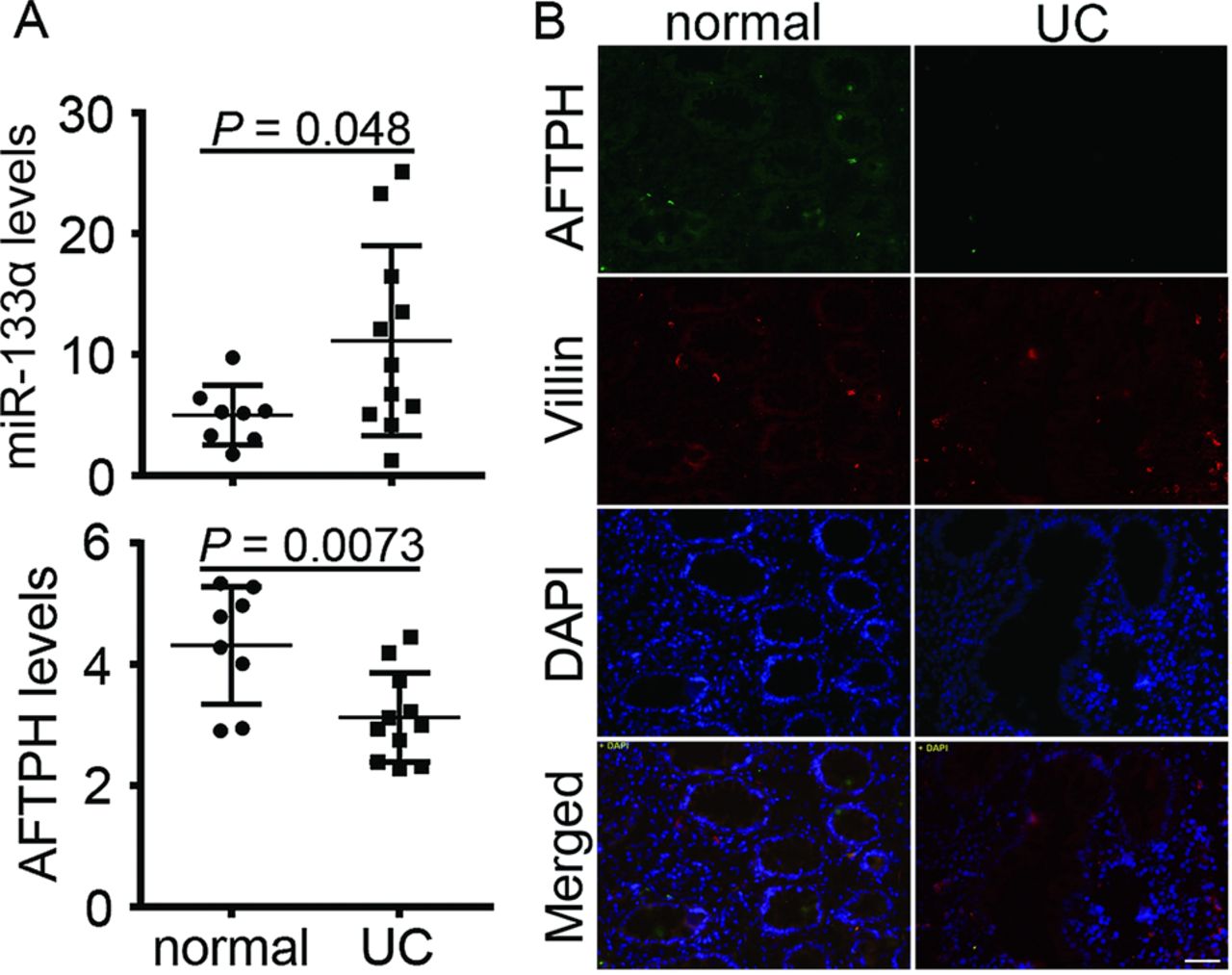
To confirm our mouse findings, we compared levels of miR-133α and AFTPH in colonic tissue samples from UC and normal colon tissues. Levels of miR-133α were significantly increased by 2.6±1.82-fold (p=0.048, figure 8A) in patients with UC with active disease, while AFTPH mRNA levels were decreased by 0.72±0.17-fold (p=0.0073, figure 8A) in the same patients. By contrast, levels of miR-133α obtained from patients with CD did not show significant difference compared with normal controls (n=8 per group, data not shown). Additionally, immunohistochemistry of colon tissues from patients with UC showed reduced AFTPH expression in the colonic mucosa when compared with normal colon tissues (figure 8B), with AFTPH expressed primarily in colonic epithelial cells, and colocalised with villin, an epithelial cell marker (figure 8B). The above results suggested that as in mouse colitis, miR-133α and AFTPH expression are inversely dysregulated in patients with UC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of miR-133α and aftiphilin (AFTPH) were significantly dysregulated in patients with ulcerative colitis (UC). (A) Levels of miR-133α and AFTPH in colon tissues from normal and patients with UC were analysed using qPCR analysis. (B) Representative images from immunohistochemistry against AFTPH and villin of colon tissues from normal and patients with UC. Scale: 50 μm.
Discussion
Coupling of NT to its high-affinity receptor NTR1 triggers MAPK38 ,39 and NF-κB7 ,40 ,41 signalling and promotes intestinal inflammation.2 ,5 ,13 We have previously shown that NT alters expression of several microRNAs, including miR-133α in human colonic epithelial cells, and presented evidence that microRNAs represent an important functional component of the effects of NT in the intestine.10 In this current study, we examined the role of miR-133α in the NT-associated intestinal inflammatory circuit in vitro and in vivo. Our results demonstrate that expression of miR-133α is increased following NT stimulation of colonocytes in vitro and normal mouse colon in vivo. We also present strong evidence that miR-133α is involved in the inflammatory cascade activated by NT/NTR1 interactions in human colonocytes, and that silencing of endogenous miR-133α in the colon diminishes acute experimental colitis in two different mouse models. Last, our results point to AFTPH as a novel downstream target of miR-133α that mediates intestinal proinflammatory signalling and cytokine transcription following NT-miR-133α interactions. Thus, the NT-regulated miR-133α and its target AFTPH may represent new targets for colonic inflammatory responses in vivo.
Our present study focused on understanding the role of miR-133α in NT-related colonic inflammation. NT/NTR1 signalling promotes acute colitis,2 ,5 while NTR1 deficiency reduces the severity of experimental colitis.5 ,13 Additionally, NT and NTR1 are overexpressed in colon tissues from patients with UC,6 and as shown in our current study, NTR1KO mice have reduced miR-133α colonic levels compared to wild-type mice. On the other hand, miR-133α and its downstream target expression have been associated with different disease states, such as colorectal cancer,21–28 myocyte development, and hypertrophy,42–44 and fibrosis in the cardiac muscle and liver.45 ,46 However, its role in colonic inflammation has never been studied. Several studies have shown that miR-133α modulates ERK29 ,30 and PI3 K/Akt21 ,31 signalling pathways, or directly targets molecules involved in these pathways.22 Consistent with the above findings, we have shown that miR-133α modulates ERK1/2 and p38 activation upon NT stimulation. We also demonstrate for the first time that miR-133α directly regulates NF-κB activation and NT exposure, together with miR-133α overexpression, in cells increased the production of proinflammatory IL-1β and suppressed that of the anti-inflammatory IL-1ra. IL-1ra antagonises IL-1β in colitis in vivo.47 ,48 Interestingly, a reduced IL-1ra/IL-1β ratio is considered as an important factor for the severity of inflammation in patients with IBD.49 Taken together, our results suggest that NT/NTR1 coupling triggers proinflammatory signalling pathways by modulating miR-133α levels in human colonic epithelial cells.
Our results showed that miR-133α levels were significantly increased in the colonic mucosa of mice with acute TNBS-induced and DSS-induced colitis, as well as colon tissues of patients with UC with active disease, compared to controls. MiR-133α was primarily expressed in colonic epithelial cells as shown by in situ hybridisation in mouse colon, with increased expression of this miR during TNBS-induced colitis. However, miR-133α was not detectable in sera from patients with UC and their normal counterparts (data not shown). To study the role of endogenous miR-133α in colitis, we introduced as-miR-133α intracolonicaly in the two experimental colitis models to avoid perturbation of normal cellular functions of non-target tissues. Our results showed that mice receiving intracolonic as-miR-133α had reduced endogenous miR-133α levels, improved mucosa histology and reduced proinflammatory cytokine expression in colon tissues of mice with colitis. Intracolonic delivery of antisense oligonucleotides is largely confined to colonic epithelial cells in vivo,50 our results suggest that miR-133α levels in colonic epithelial cells can influence the pathophysiology of colitis. However, since expression of miR-133α has also been identified in circulating monocytes,51 and in subepithelial cells of the colonic mucosa, participation of miR-133α in inflammatory signalling in colonic lamina propria cell types cannot be excluded.
We identified a novel miR-133α target, AFTPH, in which its expression level is regulated by NT/NTR1 interactions. Several pieces of evidence support our identification of AFTPH as a miR-133α downstream target. Incubation of human colonic epithelial cells with NT decreases AFTPH mRNA levels, as well as AFTPH 3′UTR-regulated luciferase activity. Most importantly, expression of AFTPH with a deleted miR-133α binding sequence on its 3′ UTR is no longer downregulated in response to NT exposure. The presence of AFTPH has not been previously recognised in the intestine, and its cellular function is not well characterised. AFTPH is essential to intracellular trafficking with binding sites for clathrin and adaptor proteins (AP-1, AP-2).52 AFTPH is localised in trans-golgi network (TGN),53 and its gene silencing leads to unregulated exocytosis of Weibel-Palade bodies in endothelial cells54 without altering TGN morphology.55 However, the role of AFTPH or TGN in proinflammatory signalling cascades has not been investigated. Here, we demonstrate that AFTPH levels are significantly downregulated in colon tissues from patients with UC, as well as in the colon of mice with TNBS-induced and DSS-induced colitis. At the cellular level, AFTPH gene silencing activates the NF-κB pathway and promotes proinflammatory cytokine IL-1β production in vitro. NF-κB activation is involved in IL-1β expression56 and administration of NF-κB inhibitor reduce IL-1β production and ameliorates colonic inflammation in mice.57 Thus, dysregulated AFTPH expression may modulate proinflammatory responses, representing a novel regulator in proinflammatory signalling pathways and colitis development. Moreover, measurements of its expression levels, together with measurements of the levels of miR-133α, may serve as biomarkers in UC.
Overall, our results represent a previously unrecognised paradigm whereby activation of a neuropeptide receptor (NTR1) in the colonic mucosa stimulates the expression of a miR (miR-133α) that modulates the development of colitis. The therapeutic potential of miR-133α silencing and stabilisation of colonic AFTPH levels against colitis deserves further investigation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Contributors Overall project design and hypotheses were developed by IKML and CP. IKML conducted experiments, analysed the data and wrote the manuscript. KB generated the human colon epithelial cells overexpressing NTR1, NCM460-NTR1. DH and AO collected and provided sera from UC and control patients and for analysing these new results now included in the revised manuscript. CP, DI and CP contributed to experimental design and provided animals and reagents. All authors participated in revising the manuscript and agreed to the final version. CP and DI supervised the overall project.
Funding This work was supported by NIH grant DK60729 (CP), the Neuroendocrine Assay Core, NIDDK P50 DK 64539 (CP), the Blinder Research Foundation for Crohn's Disease (IKML), and the Crohn's and Colitis Foundation of America (KB).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.