Article Text

Abstract
Background A major cause of enteric infection, Gram-negative pathogenic bacteria activate mucosal inflammation through lipopolysaccharide (LPS) binding to intestinal toll-like receptor 4 (TLR4). Breast feeding lowers risk of disease, and human milk modulates inflammation.
Objective This study tested whether human milk oligosaccharides (HMOSs) influence pathogenic Escherichia coli-induced interleukin (IL)-8 release by intestinal epithelial cells (IECs), identified specific proinflammatory signalling molecules modulated by HMOSs, specified the active HMOS and determined its mechanism of action.
Methods Models of inflammation were IECs invaded by type 1 pili enterotoxigenic E. coli (ETEC) in vitro: T84 modelled mature, and H4 modelled immature IECs. LPS-induced signalling molecules co-varying with IL-8 release in the presence or absence of HMOSs were identified. Knockdown and overexpression verified signalling mediators. The oligosaccharide responsible for altered signalling was identified.
Results HMOSs attenuated LPS-dependent induction of IL-8 caused by ETEC, uropathogenic E. coli, and adherent-invasive E. coli (AIEC) infection, and suppressed CD14 transcription and translation. CD14 knockdown recapitulated HMOS-induced attenuation. Overexpression of CD14 increased the inflammatory response to ETEC and sensitivity to inhibition by HMOSs. 2′-fucosyllactose (2′-FL), at milk concentrations, displayed equivalent ability as total HMOSs to suppress CD14 expression, and protected AIEC-infected mice.
Conclusions HMOSs and 2′-FL directly inhibit LPS-mediated inflammation during ETEC invasion of T84 and H4 IECs through attenuation of CD14 induction. CD14 expression mediates LPS-TLR4 stimulation of portions of the ‘macrophage migration inhibitory factors’ inflammatory pathway via suppressors of cytokine signalling 2/signal transducer and activator of transcription 3/NF-κB. HMOS direct inhibition of inflammation supports its functioning as an innate immune system whereby the mother protects her vulnerable neonate through her milk. 2′-FL, a principal HMOS, quenches inflammatory signalling.
- OLIGOSACCHARIDES
- MUCOSAL IMMUNOLOGY
- INFLAMMATION
- BREAST MILK
- BACTERIAL PATHOGENESIS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Major causes of enteric infection, Gram-negative bacteria, activate mucosal inflammation through lipopolysaccharide (LPS) binding to intestinal toll-like receptor 4 (TLR4).
Breast feeding lowers risk of disease, and human milk modulates inflammation.
Oligosaccharides are collectively the third largest components of human milk.
What are the new findings?
Human milk oligosaccharides (HMOSs) attenuate LPS-dependent induction of interleukin (IL)-8 caused by enterotoxigenic Escherichia coli , uropathogenic E. coli and adherent-invasive E. coli infection.
CD14 levels mediate LPS-dependent IL-8 inflammatory response.
HMOSs suppress CD14 mRNA transcription and CD14 translation and trafficking.
2′-fucosyllactose (2′-FL) upregulates suppressors of cytokine signalling 2 and phosphorylation of signal transducer and activator of transcription 3, and suppresses CD14 expression and release of IL-8, accounting for total HMOS activity in these infection models.
How might it impact on clinical practice in the foreseeable future?
2′-FL directly attenuating inflammation further confirms HMOSs as an innate immune system of human milk whereby the mother protects her vulnerable neonate; this supports universal breast feeding as a standard of care.
2′-FL may represent a novel oral prophylactic and therapeutic agent to quench mucosal inflammation associated with diverse inflammatory disorders of the mucosa.
Introduction
Breastfed infants have lower risk of inflammatory conditions than those fed artificially.1 Human milk suppresses inflammatory processes in vitro, and anti-inflammatory components have been documented,2 including the oligosaccharide fraction (human milk oligosaccharide, HMOS), a heterogeneous mixture of complex carbohydrate structures.3 HMOSs are comprised of at least 200 individual oligosaccharides belonging to 12 different core structural groups with lactose at the reducing end, and consist of glucose, galactose, N-acetyl-glucosamine, fucose and sialic acid.4–6 We hypothesise that HMOSs constitute an innate immune system of human milk whereby breastfeeding mothers protect their infants from enteric pathogens through three distinct mechanisms: inhibiting pathogen adhesion, prebiotic enhancement of mutualist microbiota and immunomodulation.3 ,7
Inhibition of pathogen adhesion emerges from structural homology between HMOSs and mucosal cell surface glycans. Being soluble epithelial cell membrane receptor analogues, HMOSs competitively inhibit binding of enteric pathogens to epithelial cell membranes.3 ,8 As functional prebiotics, HMOSs promote the growth of beneficial mutualist bacteria, such as Bifidobacterium bifidum, thereby influencing the colonisation and ultimate composition of intestinal microbiota.5 ,9 ,10 HMOS preparations stimulate immunomodulatory activity on the neonatal intestinal mucosal surface9 ,11–13 and modulate cytokine production.14–18 The relative activities of individual oligosaccharides and their specific mechanisms of modulating individual pathways have not been defined.
Colostrum HMOSs modulate toll-like receptor 3 (TLR3), TLR5 and interleukin 1β (IL-1b) dependent pathogen-associated molecular patterns (PAMPs) signalling pathways.18 Recognition of PAMPs by pattern recognition receptors (PRRs), including TLRs and nod-like receptors,19 ,20 is a primary mechanism by which intestinal epithelial cells (IECs) interact with the microbiota, including pathogens.21–23 When encountering microbe-specific ligands, PRRs initiate signal transduction pathways, which activate downstream nuclear transcription factors that modulate control elements of specific genes to induce transcription of proinflammatory cytokines and chemokines; their expression governs innate and adaptive responses to foreign microbes.19 ,20
The outermost membrane of Gram-negative bacteria contains lipopolysaccharides (LPS), a glycolipid PAMP.24 Pathogen-associated LPS strongly elicits several immediate proinflammatory responses in mammalian cells.25 ,26 The LPS receptor is a complex comprised of LPS-binding protein, MD2, CD14 and TLR4.27 Thus, CD14 is essential for the LPS recognition and binding that induce inflammatory signaling cascades.28 Generally, CD14 is anchored to the cell membrane by its glycosylphosphatidylinositol (GPI) moiety.29 Upon LPS recognition, TLR4s oligomerise and recruit proteins containing TIR (toll-IL-1 receptor) domains, such as myeloid differentiation primary response gene 88 (MyD88),25 promoting cascades that induce NF-κB activation and translocation to the nucleus, ultimately inducing cytokine expression.30 In most circumstances, this reaction to LPS is adaptive: During early colonisation, and during maintenance of mature microbiota, it is a component of mucosal barrier defence by restraining native microbiota to the lumen of the gut.31 This primary response to pathogen invasion thwarts pathogen passage into the body and helps clear invading microbial organisms.25 Conversely, the induction of excessive inflammation by LPS is a major component of the pathogenesis of enteropathogen infection. For example, invasion by type 1 pili Escherichia coli, such as uropathogenic E. coli (UPEC) and enterotoxigenic E. coli (ETEC), induces LPS-dependent proinflammatory IL-8 secretion.32 An abnormal TLR4-dependent reaction to LPS may underlie the pathogenesis of IBD.33
This study tested the hypothesis that HMOSs suppress LPS-induced inflammation in IECs infected by type 1 pili E. coli, measured as attenuated IL-8 secretion in ETEC-infected T84 cells and adherent-invasive E. coli (AIEC)-infected mice. The activities of individual oligosaccharides were assessed, and the mechanism of inhibition was identified through construction of a signalling pathway map.
Materials and methods
Isolation of HMOSs
HMOSs were prepared from pooled human milk,18 and endotoxin reduced to 0.01 LAL units (Genscript USA, Piscataway, New Jersey, USA) by a polymyxin B column. Pure 2′-fucosyllactose (2′-FL), 3-FL and lacto-N-fucopentaose I (LNFP I) were from Glycosyn (Waltham, MA). 3′-sialyllactose (3′-SL) and 6′-SL were from Carbosyth,(Compton, UK). Trifucosyl (1,2-1,2-1,3)-iso-lacto-N-octoase (TFiLNO) was from Glycoseparations (Moscow, Russia).
Cell lines and bacteria
IEC strains (T84/HCT8/FHs74) and HeLa cells were from ATCC (Manassas, Virginia, USA). T84 and HeLa cells were cultured in DMEM/F12 medium (Invitrogen, Grand Island, New York, USA) containing 10% FBS (Atlas Biologicals, Fort Collins, Colorado, USA). H4 cells from Dr Lei Lu (Massachusetts General Hospital), were cultured in DMEM/F12 containing 10% FBS and 30 ng/mL EGF. HCT8 was cultured in RPMI 1640 (Sigma, St. Louis, Missouri, USA) containing 10% FBS. FHs74 was cultured in X-46 medium (ATCC) containing 10% FBS and 30 ng/mL EGF. All cells were cultured at 37°C in a water-saturated atmosphere of 5% CO2.
ETEC (H10407) was purchased from ATCC. AIEC (LF82) was from Dr Jakob Møller-Jensen (University of Southern Denmark). UPEC (CF073) was from Dr Harry Mobley (University of Michigan Medical School). Salmonella enterica serovar Typhimurium (SL1344) was provided by Dr Bobby J Cherayil (Massachusetts General Hospital and Harvard). Bacteria were cultured at 37°C in Difco nutrient medium (BD Biosciences, San Jose, California, USA) at 225 rpm.
LPS stimulation in vitro
IECs were cultured at 5×104 cells per well (subconfluent) in 24-well plates (Corning life Sciences, Tewksbury, Massachusetts, USA) for 48 h in 500 μL media containing HMOS approximating physiological levels (HMOSs, 5 mg/mL; 2′-FL, 2 mg/mL; 3-FL, 0.4 mg/mL; 6′-SL, 0.5 mg/mL; 3′-SL, 0.5 mg/mL; LNFP I, 2.5 mg/mL; TFiLNO, 3 pg/mL), followed by LPS (E. coli, Sigma, St. Louis, Missouri, USA) stimulation (T84 cells 100 μg/mL; H4 cells 200 ng/mL) for 16 h. Supernatants were stored at −20°C until analysis.
ETEC stimulation in vitro
IECs were seeded at 5×104 cells per well and cultured for 48 h in 500 μL of fresh media containing 5 mg/mL HMOSs. Cells were washed with PBS twice and provided with fresh antibiotic-free, milk oligosaccharide-free media. IECs were inoculated with suspensions of ETEC with a multiplicity of infection (MOI) of 20, and incubated for 1 h. From these, the number of invading bacteria (i), the number of adherent bacteria (a) and the inflammatory response of the IECs were measured as described in online supplementary material.34–36
Inhibition of ETEC infection by cytochalasin D
Cytochalasin D, an inhibitor of actin polymerisation, inactivates host cell machinery needed for invasion by bacteria. Cytochalasin D (2 μM) was added to media of T84 cells for 30 min before ETEC inoculation (MOI=20) to inhibit invasion, and after exposure to ETEC, the ability of 2′-FL to inhibit IL-8 expression was measured in six replicate experiments.
IL-8 ELISA
The inflammatory response of T84 or H4 IECs after LPS stimulation or bacterial invasion was measured as IL-8 by ELISA (R&D Systems, Minneapolis, Minnesota, USA) in defrosted supernatants that had been centrifuged for 5 min at 4°C and 15 700×g. The colour generated at 450 nm was measured on a Versa Max plate reader (Molecular Devices, Sunnyvale, California, USA). Values were normalised to cell number (by alamarBlue).
Quantitative RT-PCR
T84 cells treated with milk oligosaccharides were stimulated with LPS or ETEC, and simultaneously analysed for CD14 protein and mRNA, as described in online supplementary material, to increase the concordance between these two types of data.
Flow cytometry analysis
Cell suspensions from T84 cells (5×105 cells/well) treated with or without HMOSs were incubated with PE-Cy7 labelled mouse antihuman CD14 mAb (BD Biosciences, San Diego, California, USA) on ice for 30 min. PE-Cy7 conjugated mouse IgGκ isotype-matched (IgG1) antibodies (BD Biosciences, San Diego, California, USA) were controls. Fluorescence was by FACScan (BD Biosciences, San Diego, California, USA), 20 000 live cells per data point, and data analysed by Accuri C6 flow cytometer software (BD Biosciences, San Diego, California, USA).
Western blots
Western blot analyses were performed as described in online supplementary material.
CD14 knockdown in IECs
To knock down CD14 translation, CD14 shRNA was transfected into T84 cells using a pRS lentiviral expression vector (OriGene Technologies, Rockville, Maryland, USA). Negative controls were vector only and vector with scrambled shRNA. Suppression of CD14 was assessed by western blot analysis 72 h post plasmid transfection.
Overexpression CD14 in HeLa cells
Overexpression of CD14 is toxic to T84, HCT8, FHs74 and H4 human IEC lines, but not to HeLa cells, whose intrinsic expression of CD14 is low. HeLa cells (2.5×106) were transfected with the pCDNA3-huCD14 plasmid by Lipofectamine 2000 for 24 h. pCDNA3 blank vector (1 μg) was a control. Overexpression of CD14 was assessed by western blot 48 h after transfection, and expressed as fold induction relative to β-actin.
Antibody arrays
T84 cells were treated with 2′-FL. Proteins were extracted, labelled, hybridised to antibody array slides and scanned. Data were analysed by ScanArrayGx/ProScanarray software, and proteins whose expression was significantly altered by the 2′-FL treatment were clustered according to signalling regulation networks by Metacore software as described in online supplementary material.
In vivo study
A murine model of AIEC infection.37 ,38 was adapted and validated. Eight-week-old female C57BL/6 mice (Charles River Laboratories) received 0.25% dextran sodium sulfate (DSS) in their drinking water for 3 days, and were given 20 mg of streptomycin by gavage on day 4; half also received 100 mg of 2′-FL in 200 μL by gavage for each of the 4 days. On the 5th day, the two groups of experimental mice were inoculated with 109 colony forming unit (CFU) AIEC via 200 μL gavage and sacrificed after an additional 4 days; a control group received DSS and antibiotic, but only a sham PBS inoculation. Body weight was monitored daily. AIEC in faeces and colonic tissue were quantified as CFU on erythromycin/ampicillin LB plates.37 Formalin (4%) fixed, paraffin-embedded 5 μm sections of mouse colon were stained with H&E. Cryosections (5 μm) of mouse colons stained with CD14 or O83 antibodies were studied by confocal microscopy. Total RNA was extracted from other colonic samples with Trizol for real-time quantitative PCR of CD14 mRNA levels, and protein was extracted for ELISA analysis of proinflammatory cytokines.
Statistical analysis
Data are presented as mean±SEM; the significance of differences was determined by post hoc analysis of variance with Bonferroni correction, (Prism software; GraphPad Software, San Diego, California, USA). Array data was analysed by GenePix Pro software. p Values of 0.05 or less are considered statistically significant. Western blot figures and photomicrographs are representative of at least three concordant independent experiments.
Results
HMOSs inhibit ETEC invasion and IL-8 secretion
Cells pretreated with HMOSs displayed reduced ETEC adherence and invasion (see online supplementary figure S1A,B), and IL-8 release (see online supplementary figure S1C) in a time-dependent manner (see online supplementary figure S1D). Higher concentrations could not compensate for shorter incubation (not shown).
HMOSs suppress CD14 expression
Of CD14, TLR4, MyD88, NF-κB and IL-8, the most pronounced attenuation by HMOSs was CD14 mRNA (see online supplementary figure S2A) and CD14 protein expression (see online supplementary figure S2B, western blot). Cell-surface CD14 was also reduced (see online supplementary figure S2C). Both processes were inhibited by HMOS irrespective of the elevated signalling during ETEC infection (see online supplementary figure S2A,B).
T84 cells exhibit high endogenous CD14 expression (see online supplementary figure S3A), that is knocked down by shRNA transfection (see online supplementary figure S3B), decreasing IL-8 induction (see online supplementary figure S3C). Knockdown of CD14 diminished the ability of HMOSs to inhibit the IL-8 induction (see online supplementary figure S3C), demonstrating that HMOS attenuation of inflammation depends upon CD14 expression.
HeLa cells express low intrinsic levels of CD14 (see online supplementary figure S3A) whose overexpression is induced by transfection with pCDNA3-huCD14 plasmids (see online supplementary figure S4B). In untreated HeLa cells, increased IL-8 secretion caused by ETEC infection is presumably mediated by factors other than CD14, and HMOSs did not inhibit this IL-8 induction (see online supplementary figure S4A). In contrast, overexpressing CD14 resulted in HMOSs suppressing ETEC-induced IL-8 secretion (p<0.01) (see online supplementary figure S4C). Accordingly, CD14 induction, the presumed target of HMOSs, was used to differentiate activities of individual oligosaccharides.
2′-FL suppresses CD14 expression
LPS mediates proinflammatory signalling during UPEC invasion of bladder epithelium32 and ETEC invasion of T84 cells (not shown). LPS at 100 μg/mL triggered significant release of IL-8 from T84 cells (see online supplementary figure S5) and HMOSs attenuated this induction (figure 1A). In this more facile simplified model, 2′-FL, 3-FL, 6′-SL, 3′-SL, LNFP I and TFiLNO (figure 1B) were tested individually at their concentrations in human milk39–41 for ability to attenuate inflammation (figure 1C). Only 2′-FL suppressed LPS-induced induction of IL-8 levels in T84 cells (figure 1B). At 2 mg/mL, 2′-FL decreased the IL-8 secretion induced in LPS-treated T84 cells by 45%, similar to the 50% inhibition by 5 mg/mL HMOSs (figure 1B). Inhibition by 2′-FL is dose-dependent (figure 1D, E) and plateaus at 80% inhibition of IL-8 induction by 4 mg/mL 2′-FL (figure 1E). Cytochalasin D inhibits the intracellular actin machinery in T84 cells that ETEC use for invasion. Incubation with 2 μM cytochalasin D did not affect basal expression of IL-8 in the presence or absence of 2′-FL (figure 1F, bars 5 and 6). Cytochalasin D pretreatment of T84 cells before ETEC infection reduced induction of IL-8 after exposure to ETEC (figure 1F, bars 3 and 7). The inability of 2′-FL to inhibit residual IL-8 expression (figure 1F, bars 7 and 8), indicates that 2′-FL inhibits only the IL-8 induced specifically by ETEC invasion.

Lipopolysaccharide (LPS) induction of interleukin (IL)-8 is inhibited by 2′-fucosyllactose (2′-FL). T84 cells were treated with human milk oligosaccharide (HMOS) or its individual components for 48 hours and then challenged by LPS. The magnitude of the inflammatory response was measured as IL-8 levels; (A), (B) and (D) are normalised to untreated LPS response (100%) and (F) to untreated enterotoxigenic E. coli (ETEC) infection to facilitate comparison across independent experiments. (A) LPS-induced IL-8 secretion was inhibited by HMOSs. (B) Isolated oligosaccharides of the HMOS mixture were tested individually at physiologically relevant concentrations for their ability to inhibit LPS induced IL-8 secretion. Schematic structures of the individual oligosaccharides are above each bar of the histogram: black circle, D-glucose; grey circle, D-galactose; black square, N-acetylglucosamine; triangle, L-fucose; dark grey diamond, N-acetyl-neuraminic acid. (C) Concentrations and inhibitory activities of the oligosaccharides 2′-FL, 2′-fucosyllactose; 3-FL, 3-fucosyllactose; 6′-SL, α2,6 sialyllactose; 3′-SL, α2,3 sialyllactose; lacto-N-fucopentaose I (LNFP I), Lacto-N-fucopentaose I; TFiLNO, trifucosyl(α1–2; α1–2; α1–3)-iso-lacto-N-octaose. (D and E) 2′-FL inhibition of IL-8 induction was dose dependent through 4 mg/mL. (F) 2′-FL specifically inhibits the IL-8 induction by ETEC invasion. Incubation of T84 cells with 2′-FL (4 mg/mL) for 48 hrs did not influence the basal expression of IL-8 (bars 1 and 2), but attenuated the ETEC induced IL-8 upregulation (bars 3 and 4). Incubation with cytochalasin D (2 μM) did not affect the basal expression of IL-8 in the presence or absence of 2′-FL (bars 5 and 6). Cytochalasin D treatment of T84 cells for 30 minutes before ETEC infection (MOI=20) to inhibit invasion reduced induction of IL-8 after exposure to ETEC (bars 3 and 7). In these T84 cells whose invasion by ETEC was blocked by cytochalasin D, the residual IL-8 expression was not changed by treatment with 2′-FL (bars 7 and 8), indicating that 2′-FL inhibited only the IL-8 induced by ETEC invasion. Mean±SEM, n=6 for all experiments; *p≤0.05; **p≤0.01; ***p≤0.001 by analysis of variance. MOI, multiplicity of infection.
At 2 mg/mL, the nominal concentration in most human milk, 2′-FL suppressed CD14 mRNA (figure 2A) and reduced cell-associated CD14 protein expression (figure 2B). Conversely, concentrations of soluble CD14 increased in the cell supernatants (figure 2C), and membrane-associated CD14 relocated to the cytoplasm (confocal microscopy) in 2′-FL-treated cells (figure 2D), for basal (p<0.05) and LPS-treated cells (p<0.05). Thus, 2′-FL, the major HMOS, accounts for the ability of HMOSs to suppress cell-associated CD14 expression and to attenuate LPS stimulated IL-8 secretion in T84 cells.

2′-fucosyllactose (2′-FL) alters CD14 expression and distribution in T84 cells. T84 cells were treated with 2′-FL for 48 h and then challenged by 100 μg/mL lipopolysaccharide (LPS). Changes induced by 2′-FL of CD14 mRNA levels, protein levels and intracellular distribution were measured. H4 cells, a model of immature intestinal epithelial cells (IECs), are more sensitive to LPS; 200 ng/mL strongly induces interleukin (IL)-8 secretion. (A) In T84 cells treated with 2′-FL for 48 h, CD14 mRNA levels, measured by RT-PCR, decreased (n=6). (B) In T84 cells treated with 2′-FL, basal and LPS-stimulated CD14 protein expression, measured by western blot, decreased (representative of n=3). (C) After 48 h of incubation with 2′-FL, concentrations of soluble CD14 in T84 culture supernatants increased (n=6). (D) After incubation with 2′-FL, membrane-bound CD14 relocated into cytoplasm in basal and LPS-treated T84 cells (n=8). (E) H4 cells are more sensitive to LPS than T84 cells, and incubation with 2′-FL attenuates IL-8 induction more strongly, supporting clinical relevance of suppression of inflammation by human milk oligosaccharides (HMOSs) and 2′-FL in the neonate. Means±SEM n=6; *p≤0.05; **p≤0.01 by analysis of variance.
While T84 cells are considered a model for enterocytes of mature intestinal mucosa, neonates consuming human milk have an immature gut more sensitive to inflammatory agents.42 The H4 enterocyte cell line models immature intestine, and is more sensitive to LPS than T84. In H4 cells, 200 ng/mL LPS induced significant release of IL-8 (figure 2E); HMOSs mitigated these changes. Thus, in mature and immature IECs, 2′-FL, the major HMOS component, accounts for the ability of HMOSs to attenuate LPS stimulated IL-8 secretion and suppress cell-associated CD14 expression.
2′-FL ameliorates inflammation induced by bacterial invasion
Consistent with the above effect on LPS, 2 mg/mL 2′-FL inhibited invasion of ETEC in T84 cells and suppressed the associated IL-8 induction (figure 3A) comparably with the 5 mg/mL HMOS seen in online supplementary figure S1. To ensure that the inhibition displayed by 2′-FL is not idiosyncratic to the T84 cell line, 2′-FL activity was tested in HCT8 cells, an IEC human ileal cell line that is especially sensitive to ETEC invasion. In these HCT8 cells, pretreatment for 48 h with 2′-FL at 2 mg/mL inhibited ETEC invasion and attenuated the consequent IL-8 secretion (figure 3B).

Interleukin (IL)-8 stimulation in other intestinal epithelial cells (IECs) and by different type 1 pili Escherichia coli is also attenuated by 2′-fucosyllactose (2′-FL). T84 or HCT8 cells were treated with 2′-FL or human milk oligosaccharide (HMOS) for 48 h and then challenged by enterotoxigenic E. coli (ETEC), adherent-invasive E. coli (AIEC), uropathogenic E. coli (UPEC) or Salmonella enterica serovar Typhimurium; bacterial invasion and IL-8 induction were measured. (A) 2′-FL inhibited ETEC invasion and IL-8 secretion in T84 IECs (derived from a colonic carcinoma). (B) HMOSs and 2′-FL inhibited ETEC invasion and IL-8 secretion in HCT8 IECs (derived from ileal adenocarcinoma). (C) HMOSs and 2′-FL inhibit AIEC (LF82, a type I pili E. coli) invasion in T84 cells; (D) HMOSs and 2′-FL inhibit UPEC (CF073, a type I pili E. coli) invasion in T84 cells. (E) HMOSs, but not 2′-FL, inhibit Salmonella enterica serovar Typhimurium (SL1344) invasion into T84 cells. Salmonella SL1344 invasion of T84 is not dependent on type I pili; therefore, the inability of 2′-FL to inhibit its invasion is consistent with 2′-FL specifically inhibiting type I pili mediated pathophysiology. The ability of HMOS to inhibit its invasion suggests that other components of the HMOSs inhibit the type III secretion virulence factors of Salmonella. Mean±SEM, n=6 for all experiments; *p≤0.05; **p≤0.01; ***p≤0.001 by analysis of variance.
To test whether the above observations were general to other type 1 pili organisms, the ability of 2′-FL to inhibit inflammatory signalling was tested in two additional type 1 pili E. coli AIEC (strain LF82) and UPEC (strain CF073). As a control, the activity of 2′-FL was tested on invasion of T84 by Salmonella enterica serovar Typhimurium (strain SL1344), whose invasion is independent of type 1 pili, instead requiring the type III secretion system or Zipper-like or Trigger-like entry processes.43 2′-FL inhibited AIEC and UPEC invasion of T84 cells by ∼50% (figure 3C, D) and the IL-8 production by ∼25% and ∼40%, respectively (not shown). Although SL1344 invasion was inhibited by total HMOSs, it was not inhibited by 2′-FL (figure 3E). This suggests that HMOS components other than 2′-FL may inhibit other mechanisms of pathogenesis, but type 1 pili pathogenesis is specifically inhibited by 2′-FL.
2′-FL induces macrophage migration inhibitory factor signal pathways that suppress inflammation
Changes in intracellular signalling associated with 2′-FL-induced changes in CD14 expression were studied in T84 cells. Signalling molecules were measured via an array of 512 antibodies to signalling proteins. Cy5/Cy3 fluorescent signal ratios were analysed using GenePix Pro array analysis software. Filtering criteria were set as internally normalised ratios of >1.3 or <0.77 based on statistical significance with correction for multiple comparisons.44
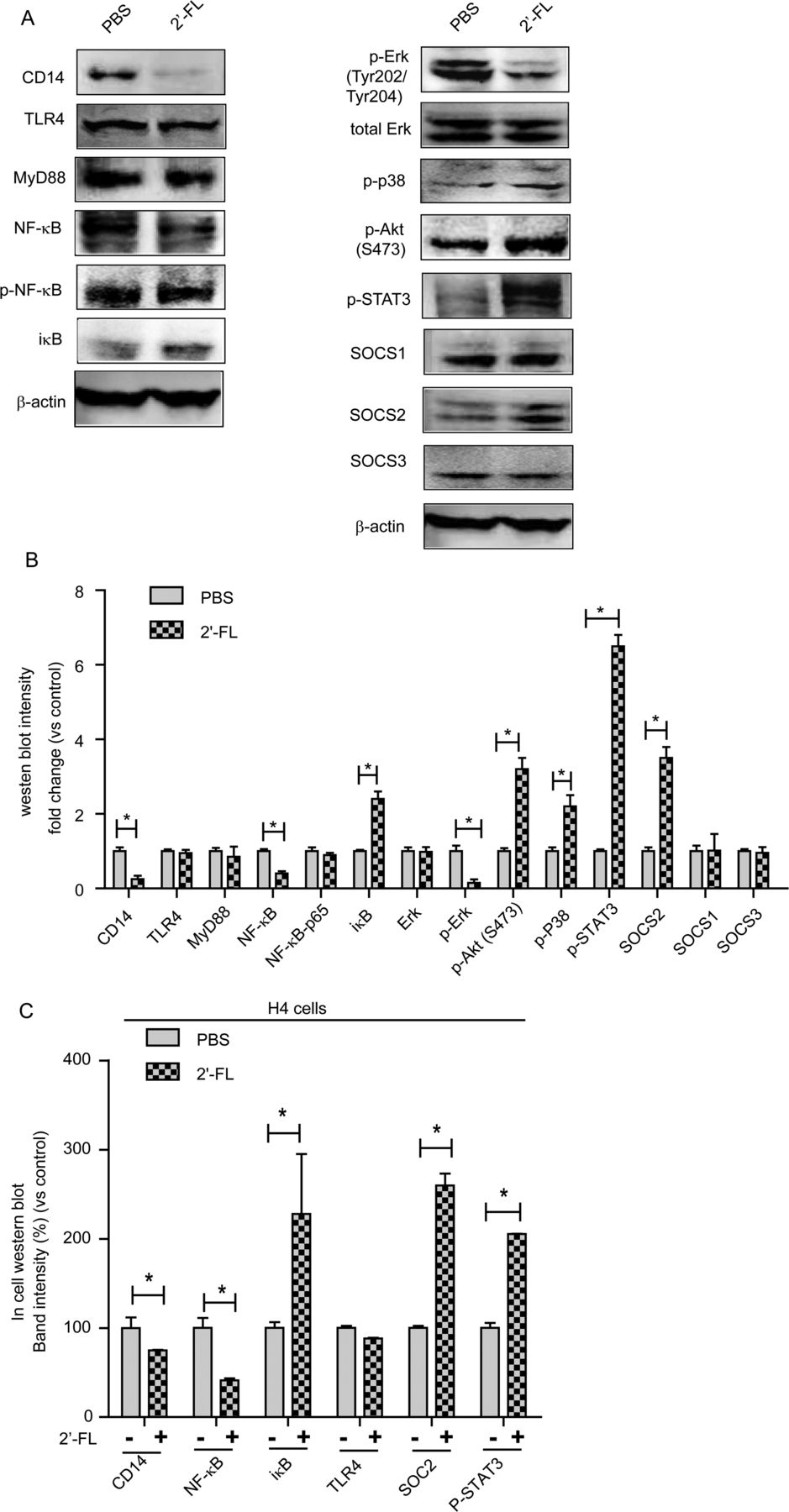
By these criteria, 2′-FL treatment of cells significantly modulated 28 signalling molecules (table 1). Functional analysis of these microarray data was conducted using integrated software from Metacore (GeneGo, http://trials.genego.com). Subsets of the macrophage migration inhibitory factor (MIF) inflammatory signalling network exhibited 2′-FL-induced changes that matched the 2′-FL-induced anti-inflammatory outcomes (see online supplementary figure S6). These changes in antibody microarray ligands induced by 2′-FL treatment were confirmed by western blot of LPS/TLR4 signal pathway mediators: 2′-FL depressed expression of CD14 and NF-κB, while inducing expression of iκB, a negative regulator of the NF-κB signal pathway (figure 4A). TLR4 and MyD88 expression was not changed (figure 4A). Erk phosphorylation decreased while p38 and Akt phosphorylation increased (figure 4A). Among the suppressors of cytokine signalling (SOCSs), 2′-FL increased expression of SOCS2 but not SOCS1 or SOCS3 (figure 4A). 2′-FL increased the phosphorylation (activation) of signal transducer and activator of transcription 3 (STAT3), a downstream signalling molecule shared by several SOCS pathways, but not of STAT1 (figure 4A). Changes in western blot intensity for all measured signalling molecules are shown in figure 4B. In H4 cells, (immature enterocyte model), 2′-FL modulated similar signal molecules: CD14 and NF-κB induction was repressed, while iκB and SOCS2 expression and STAT3 phosphorylation were induced (figure 4C). Thus, 2′-FL modulated the same signalling pathways in models of immature and mature enterocytes.
T84 cell proteins changed by 2′-FL

2′-fucosyllactose (2′-FL) treatment modifies signalling pathway intermediates. T84 cells were treated with 2′-FL for 48 h, proteins extracted, and signalling molecules measured by western blot. (A) 2′-FL modulates signalling intermediates of cytokine production in T84 cells. (B) Quantitative comparison of western blot band intensity from T84 cells (n=3). (C) H4 cells were plated at 5000 cells/well, treated with 2′-FL, and signalling proteins were measured by in-cell (in situ) western blot. H4 cell expression of signalling mediators were modulated by 2′-FL, confirming that the signalling changes observed in T84 cells are relevant to immature enterocytes (n=6). Mean±SEM; *p≤0.05 by analysis of variance.
2′-FL inhibits AIEC infection and inflammation in vivo
Three days of 0.25% DSS (a low dose) with streptomycin on the 4th day disrupts mouse microbiota. AIEC inoculation on the 5th day resulted in overt infection, manifesting as body weight loss (∼10%) 3 days and 4 days post AIEC. 2′-FL by gavage once per day for the 4 days preceding inoculation prevented the body weight loss (figure 5A). AIEC infected mice had shortened colons, but the lengths were closer to normal in AIEC inoculated mice pretreated with 2′-FL (figure 5B). Antibody against O83-antigen (expressed on LPS-positive AIEC LF82) revealed less colonisation in the 2′-FL pretreated group (figure 5C). Cultures from fresh faeces (not shown) and colon confirmed that mice in the 2′-FL pretreated group were colonised less by inoculated AIEC (figure 5D). There was less AIEC-induced CD14 expression in colonic crypts of 2′-FL treated mice, (figure 5E), fewer CD14 positive cells in muscularis mucosa (not shown), and lower CD14-mRNA levels (figure 5F). H&E staining in colon tissue of AIEC-infected mice revealed epithelial cell sloughing, immune cell infiltration, and muscularis mucosa hyperplasia, while colons of 2′-FL pretreated mice exhibited fewer of these manifestations of inflammation (see online supplementary figure S7). AIEC infection was accompanied by elevated IL-6, IL-17 and TNF-α, major inflammatory cytokines of mouse mucosa, and 2′-FL pretreatment inhibited this induction (figure 5G). IL-1β, IFN-γ and IL-10 were not significantly affected by AIEC infection nor by 2′-FL (not shown).

2′-fucosyllactose (2′-FL) modulates CD14 expression and attenuates inflammation in adherent-invasive E. coli (AIEC) infected mice. C57BL/6 mice received 0.25% dextran sodium sulfate (DSS) in their drinking water for 3 days, and were given 20 mg of streptomycin by gavage on day 4; half also received 100 mg of 2′-FL in 200 μL by gavage for each of the 4 days. On the 5th day, the two groups of experimental mice were inoculated with 109 colony forming unit (CFU) AIEC via gavage and sacrificed after 4 days; a control group received DSS and antibiotic, but only a sham PBS inoculum. (A) The body weight loss that follows AIEC infection did not occur with 2′-FL pretreatment. (B) The reduction in colon length caused by AIEC infection did not occur with 2′-FL pretreatment. (C) 2′-FL pretreatment inhibited the colonisation of the colonic mucosa by O83-positive bacteria. Representative images from six mice; orange bar=50 μm. (D) Antibiotic washed colon cultured fewer AIECs from 2′-FL pretreated mice. (E) CD14 expression was lower in colonic crypts cells of 2′-FL pretreated mice. (Confocal microscopy, orange bars = 50 μm). Images are representative of six mice. (F) CD14 mRNA levels in colon were reduced by 2′-FL pretreatment. (G) Interleukin (IL)-6, IL-17 and TNF-α levels were lower in colon of 2′-FL pretreated mice. Mean±SEM, n=6 for all groups; *p≤0.05; **p≤0.01; ***p≤0.001 by analysis of variance.
Discussion
Fucosylated moieties of the intestinal mucosa are targets for pathogens such as rotavirus A and Campylobacter jejuni.45–49 Fucosylation of the gut also provides anchors for mutualist microbes, and inability to produce these fucosylated anchors in α-1,2-fucosyltransferase 2 (FUT2) null non-secretors is associated with elevated risk of IBD, including Crohn's disease and UC, and type 1 diabetes. Indeed, the ontogeny of intestinal FUT2, which is regulated by microbiota, is requisite for gut homoeostasis.50 The presence of 2′-FL in human milk can modify the microbiota.51
In mammary gland, 2′-FL is synthesised through enzymatic transfer of fucose from guanosine 5′-diphosphate-l-fucose to lactose by FUT2.52 The resulting α-1,2-linked fucosyllactose, 2′-FL,53 is the predominant oligosaccharide in the milk of secretors, representing up to 30% of total HMOSs.54 ,55 As with all HMOSs, 2′-FL varies among individuals and over the course of lactation, and may occur in even higher concentrations in colostrum.56
Two robust biological activities have been attributed to 2′-FL: First, by binding to adhesins of Campylobacter jejuni, Salmonella typhi, Enterotoxigenic E. coli, Helicobacter pylori and to capsids of noroviruses, 2′-FL inhibits binding of these pathogens to their host receptors, the obligatory first step in their pathogenesis. Thus, 2′-FL decreases risk of diarrhoea in breastfed infants.3 ,54 ,55 ,57 ,58 Second, 2′-FL is prebiotic, a dietary carbohydrate resistant to digestion and absorption by the intestinal mucosa; in the distal gut, it stimulates growth of beneficial microbiota, including Bifidobacterium bifidum.59 A third postulated activity is attenuation of inflammation. HMOSs of colostrum quench major immunological pathways in immature human intestine ex vivo, and curb PAMP-associated stimulation. Particular pathways are attenuated by individual HMOSs,18 however specific immunomodulation by 2′-FL had not been described.
The study herein describes a major inflammatory pathway attenuated specifically by 2′-FL. The LPS dependent induction of IL-8 expression caused by invasion of IECs by type 1 pili E. coli is inhibited by 2′-FL. 2′-FL attenuates cell associated expression of CD14, thereby quenching IL-8 release. The specificity of this inhibition is illustrated by the inability of physiological concentrations of other HMOSs, including 6′-SL, 3′-SL, LNFP I and TFiLNO, or even 3-FL, a positional isomer of 2′-FL, to exhibit similar anti-inflammatory activity at their physiological concentrations (figure 1C). These results are consistent with 2′-FL modulation of CD14 expression requiring precise structural recognition by IECs. Moreover, 2′-FL inhibits only the increased IL-8 induced by infection, as evident by the inability of 2′-FL to inhibit basal IL-8 production, or IL-8 production in ETEC exposed cells in which invasion per se is inhibited. The ability of 2′-FL to inhibit inflammation associated with E. coli invasion was confirmed in AIEC-infected mice. Combined with the previous observation that galactosyl lactoses specifically inhibit polyinosinic-polycytidylic acid-induced signalling pathways,18 the data indicate that individual oligosaccharides may modulate distinct inflammatory pathways. The signalling cascade specifically modulated by 2′-FL is illustrated in figure 6. Treatment of IECs with 2′-FL increased expression of SOCS2 and phosphorylation of STAT3, and attenuated transcription and translation of cell-associated CD14, thereby quenching the typical IL-8 response of IECs to infection. 2′-FL likewise modulates IL-8 induction by exogenous LPS via identical changes in signalling.
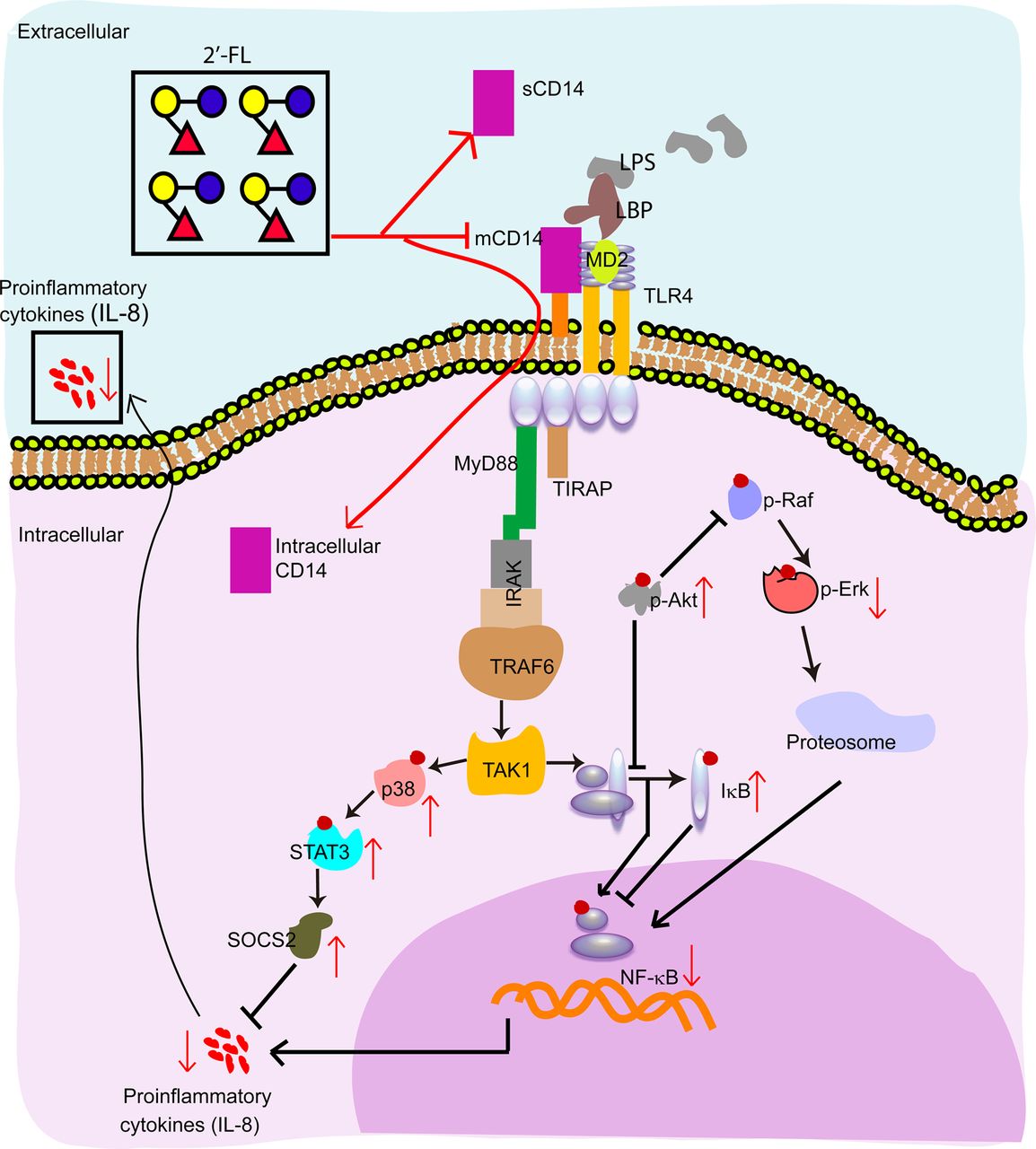
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed signalling pathway modulation by 2′-fucosyllactose (2′-FL). 2′-FL inhibits membrane-bound CD14 expression, while increasing (soluble form of CD14) sCD14 in the supernatant and the internalisation of membrane form of CD14 (mCD14). 2′-FL exposure increases the relative amounts of negative regulatory molecules, including p-Akt, p-p38, suppressors of cytokine signalling 2 (SOCS2), phosphorylated signal transducer and activator of transcription 3 (pSTAT3) and IκB, while repressing pErk and NF-κB levels. Red lines connect signalling molecules whose levels change upon exposure to 2′-FL.
CD14 plays a central role in TLR4 recognition of LPS; these three molecules form a tripartite complex that activates signalling pathways that induce production of inflammatory mediators such as IL-8.60 ,61 CD14 exists in a membrane form (mCD14) and soluble forms (sCD14),62 with each form having multiple activities. mCD14 is a GPI-anchored membrane protein62 whose expression level and location change during intestinal development and inflammatory disease.61 ,63 mCD14 is the co-receptor of LPS, and mediates cellular trafficking, such as phagocytosis of invasive bacteria and elimination of mammalian bodies during cellular apoptosis.64 ,65 Due to mCD14 being a co-receptor of LPS binding to TLR4, the reduction of mCD14 expression, such as by IL-4 and IL-13, is anti-inflammatory.61 Formation of the mCD14-TLR4 complex induces trafficking: mCD14 promotes LPS-induced endocytosis of TLR4, and mCD14 and TLR4 traffic together into the endosome.66 Internalisation of TLR4 may truncate inflammatory reactions67; this would serve to keep TLR4-mediated inflammatory responses pulsatile rather than chronic. Low levels of mCD14 reflect its loss during internalisation of TLR4, an anti-inflammatory state; conversely, the low TLR4 internalisation during inflammation would increase levels of mCD14. Note that abnormally high expression of mCD14 is associated with IBD.61 ,68
sCD14 is naturally present in normal human serum, milk and cell culture supernatants.60 ,61 ,69 sCD14 can have two forms.70 The 48 kDa sCD14 is a product of serine protease cleavage of mCD14.61 The 56 kDa sCD14 results from retention of the C-terminal sequence previously attached to GPI.61 ,71 They are considered collectively as sCD14, as the relative activities of these two forms are not clear. mCD14 is a more efficient activator of TLR4 signalling than sCD14. Endothelial cells express TLR4 but are intrinsically CD14-negative; therefore circulating sCD14 initiates TLR4 inflammatory signalling.61 Immature IECs also express little CD14 relative to that in mature cells. Therefore, sCD14 in human milk, when bound to the LPS of Gram-negative bacteria in the infant intestinal tract, is proposed to be a ‘sentinel’ molecule that activates TLR4-mediated inflammatory signalling.72 In contrast to these local proinflammatory effects, circulating sCD14 exhibits anti-inflammatory effects in tissues distant from the inflammation.73 In these mCD14 positive tissues, circulating sCD14 competes with mCD14, promotes internalisation of TLR4, thereby decreasing the TLR4 response to LPS74 and limiting the intensity and duration of the inflammatory response. The effects of 2′-FL on CD14 occur at multiple levels: 2′-FL decreases levels of cell-associated CD14 by suppression of CD14 transcription and translation (figure 2A,B); 2′-FL induces endocytosis of surface mCD14 into the cytoplasm (figure 2D); and 2′-FL increases the secretion of sCD14 to extracellular culture supernatants (figure 2C). Each of these could contribute to the quenching of inflammatory activity by 2′-FL.
Downstream TLR4-LPS-CD14 activation of several major signalling molecules was modulated by 2′-FL, summarised in figure 6. Most mediate either the Akt or the SOCS2 pathway within the MIF network. The phosphatidylinositol 3-kinase/Akt pathway limits LPS activation of signalling pathways.75 ,76 pAkt blocks pNF-κB and pRaf.75 pRaf promotes p-Erk and induces NF-κB.75 Thus, the striking upregulation of pAkt by 2′-FL represses pErk and NF-κB, an anti-inflammatory effect that is consistent with the experimental data.
SOCS proteins are negative regulators of cytokine signalling. They are activated most commonly through the janus kinase and STAT pathways.77 ,78 SOCS2 (figure 4A,C), as well as its upstream signalling molecules p-p38 and pSTAT3 (figures 4A,B and 6), are upregulated by 2′-FL. SOCS2-inhibited signalling molecules are thought to regulate growth more than immune function.79 However, the data herein, in which 2′-FL upregulates SOCS2 while quenching IL-8 production, suggest that SOCS2 may suppress mediators of LPS-induced IL-8 activation. 2′-FL stimulation of SOCS2 could also promote maturation of immature epithelial cells through signalling molecules related to proliferation (Ki-67) and cell cycle (CDC25C), an independent more indirect mechanism to attenuate inflammation. This phenomenon may be addressed in further studies.
2′-FL suppression of cell-associated CD14 expression prevents LPS-dependent type 1 pili E. coli invasion while modulating the inflammation induced by LPS at the mucosal surface. This ability of 2′-FL to quench LPS stimulated inflammation may be of clinical relevance for neonates. Relative to mature intestinal mucosa, more TLR4 is on the cell surface of the immature mucosa, and this may be linked to the hypersensitivity to inflammatory stimuli exhibited by developing intestine.42 This hypersensitivity creates a challenge during initial colonisation, where the innate immune system is naïve to all microbes. Although Gram-negative (LPS-positive) E. coli may represent an especially potent inflammatory stimulus,80 exposure to exogenous endotoxin causes IECs to acquire TLR tolerance immediately after birth. This facilitates microbial colonisation by mutualist and commensal bacteria, allowing development of a stable intestinal host-microbe mutualism81; imbalance leads to dysbiosis, increasing risk of NEC.82 If the 2′-FL prevalent in most human milk is a major factor in quenching inflammation during this transition from fetal to colonised postnatal intestinal mucosa, 2′-FL should inhibit inflammation in most IEC models.
2′-FL suppressed inflammatory signalling in two additional cell culture IEC models. HCT8 is more sensitive to inflammatory stimuli, and closer than T84 to the physiology of IECs in situ. The H4 line, derived from immature intestine, recapitulates many IEC characteristics of immature intestine. Pretreatment of either cell line with 2′-FL induced resistance to ETEC invasion and inhibition of IL-8 induction similar to that observed in T84 cells (see online supplementary figure S1, S2 and S4). The ability of 2′-FL to inhibit AIEC colonisation was confirmed in vivo, measured as lower AIEC in faeces and colon following a standard inoculum. In AIEC-infected mice, 2′-FL pretreatment prevented body weight loss, and quenched inflammation through repression of CD14 expression. AIEC induction of proinflammatory cytokine levels was prevented by 2′-FL, resulting in lower histopathology inflammatory scores and other signs of colon inflammation (figure 5).
Moreover, 2′-FL inhibits multiple type 1 pili invasive E. coli: the primary model ETEC strain, UPEC and AIEC. Type 1 pili E. coli are major pathogens causing millions of cases annually of urinary (UPEC) or gastroenterological (ETEC) infection,83 ,84 and AIEC are associated with IBD.85–88 Inhibition of E. coli pathogenesis by 2′-FL may underlie the association between breast feeding and decreased risk of disease, and suggests a novel treatment for type 1 pili E. coli-associated enteric and urinary tract infections.89 Moreover, the inhibition of salmonella infection by HMOSs but not by 2′-FL implies that HMOSs contain other oligosaccharides that protect against pathogens whose critical determinants are distinct from type 1 pili.
Breast feeding is associated with decreased risk of inflammatory infectious diseases, and 2′-FL is a principal human milk component. 2′-FL alone inhibits proinflammatory signalling induced by type 1 pili E. coli pathogen invasion (or by LPS) by suppressing CD14 expression in IECs. This quenching of inflammation by 2′-FL is additional evidence that HMOSs comprise a milk-borne innate immune system through which the mother protects her infant from environmental insult while the infant's immature mucosal immune system is developing and maturing. But this is only one oligosaccharide and one pathway; 2′-FL and other oligosaccharides of human milk could influence additional mucosal signalling pathways, thereby contributing towards the orderly maturation and colonisation of the infant gut. HMOSs may represent a fresh category of prophylactic and therapeutic agents, and 2′-FL might be an early candidate for clinical trials against LPS-mediated inflammation.
Acknowledgments
The authors thank Paul McCabe for assistance with editing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table
Footnotes
YYH and SBL contributed equally.
Contributors YYH, SBL and DSN designed the research; YYH, SBL, DEK, SL, NTL, YH, SBF and DRH conducted the experiments; YYH, SBL and DSN analysed the data; YYH and DSN wrote the first draft of the manuscript and all authors contributed to its completion.
Funding Partially supported by NIH grants R01HD059140, U01AI075563 and P01HD013021 and Abbott Nutrition.
Competing interests DSN owns stock in Glycosyn, which makes human milk oligosaccharides. This is being managed by Boston College. Other authors have no competing interests to declare.
Provenance and peer review Not commissioned; externally peer reviewed.