Article Text

Abstract
Objective To identify a causal mechanism responsible for the enhancement of insulin resistance and hyperglycaemia following periodontitis in mice fed a fat-enriched diet.
Design We set-up a unique animal model of periodontitis in C57Bl/6 female mice by infecting the periodontal tissue with specific and alive pathogens like Porphyromonas gingivalis (Pg), Fusobacterium nucleatum and Prevotella intermedia. The mice were then fed with a diabetogenic/non-obesogenic fat-enriched diet for up to 3 months. Alveolar bone loss, periodontal microbiota dysbiosis and features of glucose metabolism were quantified. Eventually, adoptive transfer of cervical (regional) and systemic immune cells was performed to demonstrate the causal role of the cervical immune system.
Results Periodontitis induced a periodontal microbiota dysbiosis without mainly affecting gut microbiota. The disease concomitantly impacted on the regional and systemic immune response impairing glucose metabolism. The transfer of cervical lymph-node cells from infected mice to naive recipients guarded against periodontitis-aggravated metabolic disease. A treatment with inactivated Pg prior to the periodontal infection induced specific antibodies against Pg and protected the mouse from periodontitis-induced dysmetabolism. Finally, a 1-month subcutaneous chronic infusion of low rates of lipopolysaccharides from Pg mimicked the impact of periodontitis on immune and metabolic parameters.
Conclusions We identified that insulin resistance in the high-fat fed mouse is enhanced by pathogen-induced periodontitis. This is caused by an adaptive immune response specifically directed against pathogens and associated with a periodontal dysbiosis.
- DIABETES MELLITUS
- DIET
- BACTERIAL INFECTION
- BACTERIAL PATHOGENESIS
- IMMUNE RESPONSE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Cross-sectional studies show a positive correlation between periodontitis and type 2 diabetes.
A diabetogenic fat-enriched diet induces periodontitis via increased periodontal pathogens and lipopolysaccharides (LPS)-signalling activation.
Bacterial members of dental plaque have been found in the microbiota of atherosclerosis plaques in humans.
A gut microbiota dysbiosis induces metabolic disease.
An adaptive immune system induces metabolic disease.
What are the new findings?
A high-fat diet (HFD) increases periodontal microbiota diversity in mice, conversely to that reported for gut microbiota.
Porphyromonadaceae abundance in periodontal microbiota is linked to insulin resistance and positively correlated to alveolar bone loss.
Periodontitis induces antibodies against Porphyromonas gingivalis (Pg), and the decrease of these specific antibodies was associated with the impaired glucose metabolism in HFD-fed mice.
Pg-LPS was shown to be responsible for the periodontitis-induced metabolic impairment under an HFD.
The modulation of the regional (cervical) adaptive immune system is causally responsible for periodontitis-induced insulin resistance.
How might it impact on clinical practice in the foreseeable future?
Increased Porphyromonadaceae abundance in the periodontal microbiota of diabetic patients may represent a new prognostic marker for periodontitis-aggravated insulin resistance.
Pg-LPS-based inhibitory therapy and antibiotics targeted directly against Porphyromonadaceae may be useful to prevent the deleterious effects of periodontitis on glucose homeostasis in diabetic patients.
An anti-inflammatory strategy directed against the regional immune system could reduce the incidence of insulin resistance and type 2 diabetes.
A vaccination strategy against Pg may reduce the impact of periodontitis on glucose metabolism.
Introduction
Type 2 diabetes (T2D) is now considered a pandemic disease. The causal origin of this accelerating development is related to several interacting factors such as sedentary lifestyle, excessive body weight (BW), stress and bad feeding habits.1 Markedly, the prevalence of periodontitis within the diabetic population is 60% while it ranges from 20% to 50% in the general population.2 ,3 In patients with periodontal diseases the incidence of pre-diabetes or undiagnosed T2D is increased by 27–53%.4 ,5 Of note, treating periodontal diseases reduced by 0.4% glycosylated haemoglobin in patients with T2D.5 Nonetheless, the causal link between periodontitis and T2D is still unknown. The last decade demonstrated that the incidence of metabolic and cardiovascular diseases6 was also linked to gut microbiota dysbiosis.7 ,8 Therefore, we reasoned that a dysbiosis of periodontal microbiota could be responsible, at least in part, for incidence of metabolic diseases,9 and that the lipopolysaccharides (LPS) from Gram-negative bacteria could be released in local and systemic organs, leading to metabolic endotoxemia and insulin resistance as described in mice10 and humans.11
Numerous Gram-negative LPS-releasing periodontal pathogens are located within the periodontal biofilm. As an example, Prevotella intermedia (Pi) induces periodontal tissue destruction12 and its prevalence is higher in patients with diabetes.13 Interestingly, periodontal bacteria, such as Fusobacterium nucleatum (Fn), have been found within the atherosclerotic plaque, suggesting a potential translocation towards the systemic circulation.14 Moreover, Porphyromonas gingivalis (Pg) is associated with chronic diseases such as cardiovascular diseases,15 rheumatoid arthritis,16 pancreatic cancer17 and non-alcoholic steatohepatitis (NASH).18 In fact, Pg is present in the injured liver and aggravates NASH via promoting inflammation.18 It has been established that metabolic diseases are characterised by a chronic low-grade inflammation named metabolic inflammation,19 where macrophages and T-lymphocytes are recruited within metabolic tissues such as liver and adipose depots and release proinflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and plasminogen activator inhibitor (PAI)-11 that impair insulin action. Hence, insulin resistance seems to be secondary to the onset of an inflammatory process,1 in which innate and adaptive immune responses may promote inflammatory reactions driven by gut microbiota.20 ,21
Altogether, these evidences suggest that a periodontal microbiota dysbiosis could initiate first a regional and then a systemic metabolic inflammation promoting insulin resistance and T2D. To demonstrate the causal role of periodontal diseases as a risk factor for T2D and the relevance of the innate and adaptive immune responses, we have set-up a unique and specific model of periodontitis by Gram-negative bacterial periodontal-pathogen colonisation in mice. Features of periodontal microbiota and glucose metabolism have been also investigated. We report the causal role of regional adaptive immune system response in the worsening of insulin resistance induced by periodontitis and precisely the LPS from Pg. An impaired specific immune response against Pg could be responsible for the enhancement of the incidence and the gravity of T2D.
Materials and methods
Animals and experimental procedures
C57Bl/6J wild-type (WT) (Charles River, L'Arbresle, France) female mice were group-housed (six mice per cage) in a specific pathogen-free controlled environment (inverted 12 h daylight cycle, light off at 10:00). Five-week-old mice were randomised into two groups: group 1 was colonised (Co) and group 2 served as control. For group 1, 1 mL of a mix of 109 colony-forming unit (CFU) of each periodontal pathogen such as Pg ATCC 33277, Fn and Pi previously identified,22 in 2% carboxymethylcellulose was applied at the surface of the mandibular molar teeth, four times a week, during 1 month. Control mice received the vehicle only. Each group was divided into two subgroups and fed with either a normal chow (NC, energy content: 12% fat, 28% protein and 60% carbohydrate; A04, Villemoisson-sur-Orge, France) or a diabetogenic, high-fat carbohydrate-free diet (HFD; energy content: 72% fat (corn oil and lard), 28% protein and <1% carbohydrate; SAFE, Augy, France) for 3 months.23 The groups were labelled as following: NC+vehicle (NC), NC+colonisation (NC-Co), HFD and HFD+colonisation (HFD-Co).
Periodontal and gut microbiota analysis
Total periodontal DNA was extracted from frozen mandibles and faeces as previously described.24 For periodontal tissue, the whole 16S bacterial DNA V2 region was targeted by the 28F-519R primers and pyrosequenced by the 454 FLX Roche technologies at Research&Testing Laboratory (http://www.researchandtesting.com/, Texas, USA). An average of 4907 sequences was generated per sample. For gut microbiota, the MiSeq technique was applied to generate an average of 10 000 sequences per sample by Research&Testing Laboratory. The cladogram in figure 1K and the linear discriminant analysis (LDA) score analysis in online supplementary figure S1 have been generated via the Galaxy website of Huttenhower's Lab.25

Oral colonisation with Porphyromonas gingivalis (Pg), Fusobacterium nucleatum (Fn) and Prevotella intermedia (Pi) induces periodontitis associated with local and systemic immune disorders. (A) Mice were colonised with Pg, Fn and Pi or by vehicle solution for 1 month and then randomised into four groups: normal chow (NC, blue bar, n=6), normal chow colonised (NC-Co, purple bar, n=6), high-fat diet (HFD red bar, n=7) and high-fat diet colonised (HFD-Co, green bar, n=10). (B) Hemi-mandible for each group, as reconstructed by micro-CT (Viva CT40; Scanco Medical, Bassersdorf, Switzerland). (C) Alveolar bone loss (yellow line) for each group. (D) Tumour necrosis factor (TNF)-α, plasminogen activator inhibitor (PAI)-1, interleukin (IL)1-β and IL-6 expression in periodontal tissue. (E) Histological examination for hemi-mandibles (PT, periodontal tissue; T, tooth) stained with H&E, F4/80, CD3 and CD20 antibodies: cells count is shown on the right side of each panel series. Arrows show infiltrated cells within the anatomical area and insets show a magnification of the slide. Number and relative abundance of immune cell types explored at 3 months in cervical lymph nodes (F and G) and in spleen (H and I) and the blood (J). (K) Linear discriminant analysis effect size (LEfSe) analysis-based cladogram for periodontal microbiota of each group. (L) Complete linkage clustering using Euclidean distance. Data (mean±SEM) and one-way analysis of variance (ANOVA) followed by Tukey's test used for *p<0.05 and ****p<0.0001 when compared to HFD, §p<0.05; §§p<0.001 §§§§p<0.0001 when compared to NC and $p<0.05 when compared to NC-Co.

Continued

Continued
Quantification of mandibular alveolar bone resorption
Hemi-mandibles were scanned using a high-resolution µCT (Viva CT40; Scanco Medical, Bassersdorf, Switzerland).26 Data were acquired at 45 keV, with a 10 μm isotropic voxel size. Six linear measurements were obtained from each molar by using a stereomicroscope with an on-screen computer-aided measurement package. The alveolar bone loss (ABL) (mm) was measured from the cemento-enamel junction to the alveolar bone crest for each molar.27 Three-dimensional reconstructions were generated from a set of 400 slices.
Real-time quantitative PCR analysis for periodontal tissue
Total RNA from periodontal tissue was extracted using the TriPure reagent (Roche, Basel, Switzerland). cDNA was synthesised using a reverse transcriptase (Applied Biosystems, Fost City, USA) from 1 µg of total RNA as previously explored.28 The primers (Eurogentec, San Diego, USA) used were (5′ to 3′): TNF-α, forward TGGGACAGTGACCTGGACTGT; reverse, TCGGAAAGCCCATTTGAGT; IL-1β, forward TCGCTCAGGGTCACAAGAAA; reverse CATCAGAGGCAAGGAGGAAAAC; PAI-1, forward ACAGCCTTTGTCATCTCAGCC; reverse CCGAACCACAAAGAGAAAGGA; and IL-6 forward ACAAGTCGGAGGCTTAATTACACAT; reverse TTGCCATTGCACAACTCTTTTC. The concentration of each mRNA was normalised for RNA loading against the ribosomal protein L19 (RPL19) (forward GAAGGTCAAAGGGAATGTGTTCA; reverse CCTTGTCTGCCTTCAGCTTGT) as an internal standard, and the data were analysed according to the 2−ΔΔCT method.28
Intraperitoneal glucose-tolerance test and in vivo euglycemic/hyperinsulinemic clamp
Six-hour fasted mice were injected with glucose into the peritoneal cavity (1 g/kg). Blood glucose was measured with a glucometer (Roche Diagnostics, Meylan, France) on 2 μL of blood collected from the tip of the tail vein at −30, 0, 30, 60 and 90 min after the glucose injection. To assess insulin sensitivity, a catheter was indwelled into the femoral vein as previously described.10 After full recovery from the surgery and 6 h of fasting, the whole-body glucose utilisation rate was evaluated in euglycemic/hyperinsulinemic conditions, as described elsewhere.10
Histological analyses
Hemi-mandibles were excised, fixed in 4% paraformaldehyde for 48 h and embedded in paraffin. Hemi-mandibles samples were cut with a microtome in the transverse direction following the main axis of tooth from coronal to apical. Then, sections (4 µm thickness) were stained with H&E. Immuno-histological analyses were performed using primary antibodies against F4/80 (AbD Serotec, Colmar, France), CD3 (Spring Bioscience, Pleasanton USA) and CD20 (Thermo Scientific, Rockford, USA), and revealed by R.T.U. (Ready-to-Use) Vectastin Elite (Vector Laboratories, Burlingame, USA) and for diaminobenzidine (DAB) by ImmPACT DAB Substrate (Vector Laboratories), to quantify the infiltration of immune cells. Slides were scanned with ‘panoramic digital scanner 250’ with Z-Stack function and the objective 40× (3DHISTECH). Cells subpopulations counting was done with the Panoramic Viewer software (3DHISTECH) and carried into the Lamina propria gingivae and periodontal ligament on average surface 0.1 mm2 on each tissue section. Five microscopic fields were counted on each slide by two independent naïve investigators.
Surface staining and antibodies treatment of immune cells from cervical lymph nodes, spleen and blood
Mononuclear cell suspensions were incubated for 15 min with anti-CD16/32 to block Fc receptors and then with antibodies, anti-CD4 APC (RMA4-5, eBioscience), CD8 V450 (53.6.7, BD Bioscience), anti-CD11b APC-eFluor780 (M1/70, eBioscience), CD45 V500 (30F11, BD Bioscience), anti-CD19 FITC (1D3, BD Bioscience) and anti-TCR PerCP-Cy5.5 (H57, eBioscience) for 30 min on ice. LIVE/DEAD Fixable Cell Stain Kit (Life Technologies) was used to remove dead cells. All data were acquired using a digital flow cytometer (LSR II Fortessa, Becton Dickinson), and analysed with FlowJo software (Tree Star).
Immunotherapy
Cervical lymph nodes were harvested either from mice colonised with bacteria mixture as described above or not colonised. Cervical lymph node cells (107 total) were injected into the peritoneal cavity from mice with periodontitis (periodontitis transfer cells) or without (healthy transfer cells) and an intraperitoneal glucose-tolerance test (IPGTT) was assessed after transfer and after colonisation with periodontal pathogens.
Immunisation
An injection of 106 CFU of Pg, Fn or Pi or the mix of the three bacteria, inactivated by oxygen-exposure during 48 h, was given in the footpad muscle. Control mice were injected with saline. Then, periodontitis was induced (as described above) 1 month after the immunisation in 3 months HFD-fed mice.
Pg-LPS treatment
Mice were subcutaneously implanted with an osmotic minipump (Alzet Model 2004; Alza, Palo Alto, California, USA) delivering either ultrapure LPS from Pg at 300µg/kg/day as previously reported10 (CAYLA-InvivoGen, Toulouse, France) or NaCl (0.9%) to ensure a continuous 28 days infusion. Then, mice were fed a NC or HFD for 1 month. Finally, mice were sacrificed by cervical dislocation and tissues were collected and snap-frozen in liquid nitrogen.
Anti-Pg antibodies measurement
Immunoglobulin G antibodies specific to LPS of Pg were measured using a home-made ELISA. The wells of 96-well flat-bottom microtiter plates were coated in triplicates with LPS of Pg. After washing and blocking the plates, serum samples were added to individual wells and specific mouse IgG antibodies were detected with an alkaline phosphatase-conjugated antimouse immunoglobulin. The absorbance was read at 405 nm using an ELISA plate reader. The results were expressed as an ELISA index (EI), which was the mean OD 405 of a given serum sample divided by the mean OD 405 of the calibrator (reference serum).29
Plasma biochemical assays
Fifty microlitres of blood were sampled from the retro-orbital sinus in awake condition in 6- h-fasted mice. For insulin, the plasma was separated and frozen at −80°C. Also, 10 µL of plasma were used to determine insulin concentration with an Elisa kit (Mercodia, Uppsala, Sweden) following the manufacturer's instructions. Plasma cytokine concentration was determined by the MILLIPLEX MAP system (Luminex, Austin 12212 Technology Blvd., Austin, Texas, USA/Merck Millipore Headquarters 290 Concord Road Billerica, Massachusetts, USA).
Statistical analysis
Results are presented as mean±SEM. One-way analysis of variance (ANOVA) followed by Tukey's post-test was used to assess inter-groups differences, except for the IPGTT, where two-way ANOVA followed by Bonferroni's post-test was applied. *p<0.05; **p<0.01; ***p<0.001 and ****p<0.0001 when compared to HFD, §p<0.05; §§p<0.001 §§§§p<0.0001 when compared to NC and $p<0.05 when compared to NC-Co defined statistical significance. Statistical analyses were performed using GraphPad Prism V.5.00 for Windows Vista (GraphPad Software, San Diego, California, USA). The dendrogram on figure 1L was drawn by PermutMatrix software;30 figure 2I (principal coordinate analysis (PCoA)) was drawn by XLSTAT for Microsoft Windows Excel.

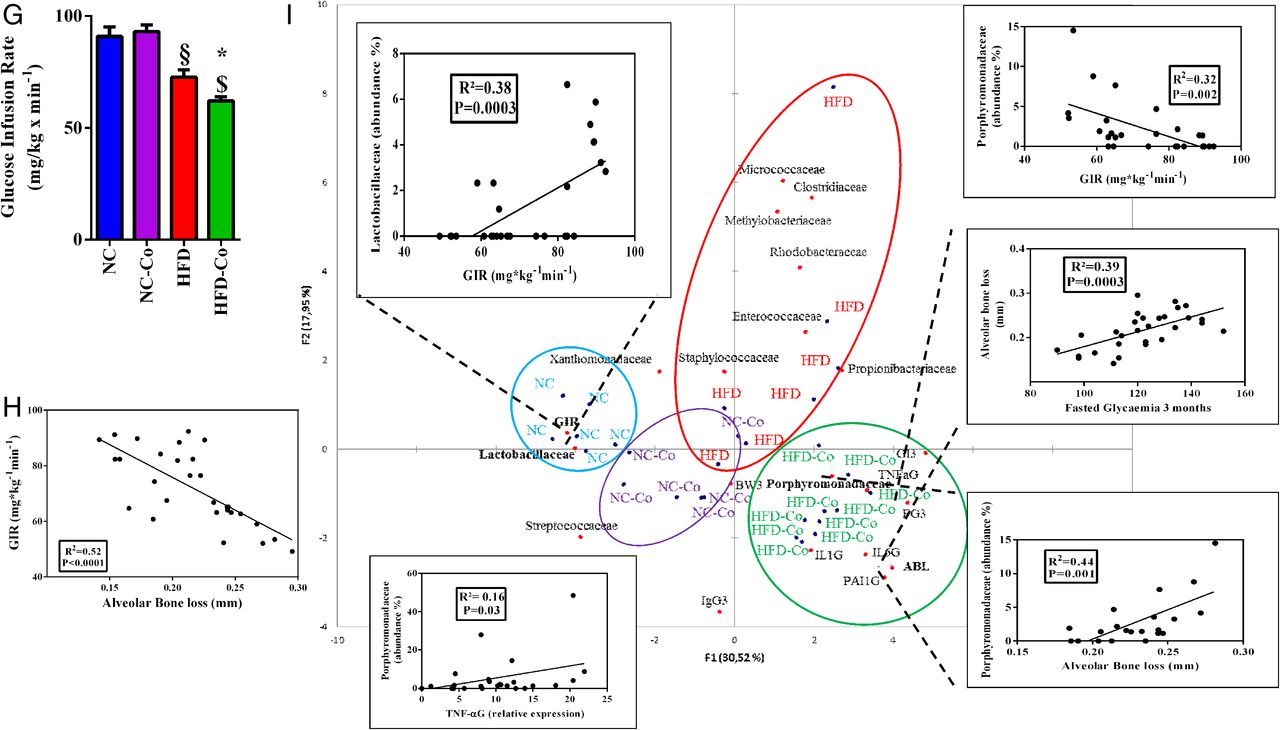
Periodontitis enhances high-fat diet (HFD)-induced metabolic disorders in mice. Glycaemic profiles (mg/dL) during an intraperitoneal glucose-tolerance test (IpGTT; normal chow (NC, blue bar, n=6), normal chow colonised (NC-Co, purple bar, n=6), high-fat diet (HFD, red bar n=7) and high-fat diet colonised (HFD-Co, green bar, n=10)) and glycaemic indexes as inset; ratio fat/lean for each group during 1 month (A and B), 2 months (C and D) and 3 months (E and F). (G) Insulin sensitivity evaluated by the euglycemic-hyperinsulinemic clamp technique. (H) Correlation between glucose infusion rate (GIR) and alveolar bone loss (ABL). (I) Principal coordinate analysis (PCoA) between dominant bacterial families from periodontal microbiota (abundance >1%, detected at least in one mouse) and metabolic parameters such as ABL, GIR, gingival inflammation (TNFaG, IL1bG, PAI1G, IL6G, where ‘G’ stands for gingival), immunoglobulin G score, glycaemic index, fasted glycaemia and body weight at 3 months (IgG3, GI3, FG3, BW3, respectively): the three insets represent the correlation between GIR and Lactobacillaceae family, Porphyromonadaceae family and Porphyromonadaceae family with ABL. Data are mean±SEM. Significant results when *p<0.05; **p<0.01 and ***p<0.001 when compared to HFD, §p<0.05 and §§§p<0.001 when compared to NC and $p<0.05 when compared to NC-Co as determined by two-way analysis of variance (ANOVA) with Bonferroni's post-test for (A), (C) and (E) and one-way ANOVA followed by Tukey's post-test for (B), (D), (F) and (G).

Continued
Results
Pg, Fn and Pi periodontal pathogens promote periodontal, cervical and systemic immune disorders together with periodontal microbiota dysbiosis during an HFD
Pg, Fn and Pi,31 periodontal pathogens, are drivers of the development of periodontitis in mice. Here, to demonstrate that periodontitis is an aggravating risk factor for diet-induced metabolic disease, we generated a unique mouse model. First, periodontitis was induced by colonising 5-week-old WT C57Bl6/J female mice with all three pathogens; then, mice were fed with an NC or a diabetogenic/not obesogenic HFD (figure 1A).
We validated our model by showing periodontal pathogens-induced mandibular ABL, a feature of periodontitis, on NC. Moreover, this parameter was worsened on HFD (figure 1B, C). Then, we studied the periodontal tissue looking for a putative inflammatory status. As shown in figure 1D, NC-fed colonised mice displayed a significant increased gene expression for all the analysed cytokines. Moreover, the pro-inflammatory effect due to TNF-α and PAI-1 was increased by HFD (figure 1D). Subsequently, given this evidence, we analysed histological sections of hemi-mandibles. We showed by H&E staining that cells infiltrated the periodontal tissue (figure1E) after the colonisation with the periodontal pathogens on NC. In addition, we characterised the cell types by immunostaining and showed an increased macrophage (cells F4/80+), lymphocyte T (cells CD3+) and lymphocyte B (cells CD20+) number in the same experimental conditions. In response to the HFD treatment, the number of cells increased when compared to NC-fed mice. Additionally, in colonised HFD-fed mice the number of immune cells even further increased over that of NC, NC-Co and HFD mice, showing the impact of the dietary treatment and of the colonisation on the inflammatory process in periodontal tissue (figure 1E).
Next, to identify whether periodontitis and local inflammation may be associated with an impaired immune system, we quantified local (cervical lymph node) and systemic (spleen) adaptive and innate immune system cells. HFD feeding increased the number of cells in both cervical lymph node and spleen when compared to NC-fed mice. Interestingly, periodontitis blunted this increase in HFD-fed mice only (figure 1F, H). In the latter, this variation was due to a strong reduction in the frequency of innate CD11b+ cells (gating on CD3-CD19-CD11c-CD11b+) in cervical lymph nodes and spleen, whereas periodontitis increased the frequency of T-lymphocyte and B-lymphocyte (CD4+, CD8+ and CD19+) during HFD only (figure 1G, I).
To further explore the systemic effect of periodontitis, we analysed immune cells in blood, where the above reported modifications were confirmed for all cell types and especially for dendritic cells (CD11b+CD11c+) and innate CD11b+CD11c− cells (figure 1J). By contrast, periodontitis had no significant impact on any cell type whatever the tissue under NC (figure 1F–J). Indeed, the periodontal colonisation increased the number of antibodies anti-Pg on NC, whereas the HFD treatment reduced IgG serum levels and antibodies anti-Pg in colonised mice only (table 1). Conversely, HFD increased IgG serum levels independently of colonisation at both 2 and 3 months of treatment (table 1). Moreover, the periodontitis increased blood IL-6 on HFD and decreased blood interferon-γ concentrations on HFD and NC at 3 months (table 1).
Systemic metabolic and inflammatory parameters in fasted mice over the time course of the treatment
Then, to evaluate whether the change in immune features was linked to a periodontal microbiota dysbiosis, we studied the composition of periodontal microbiota on both NC-fed and HFD-fed colonised mice. On NC, colonisation significantly increased the genus Lactococcus, as shown by the cladogram based on the LDA effect size (figure 1K). On HFD, colonisation significantly increased the Porphyromonadaceae family, in accordance with Pg colonisation (figure 1K). We also showed that HFD induces a periodontal microbiota dysbiosis based on the significant increase in genera Bacteroides, Clostridium and Ruminococcus. The LDA scores and the details (Phyla to Species) for all the periodontal microbiota changes are reported in online supplementary figure S1. Interestingly, periodontitis affected the overall periodontal microbiota profile of HFD-fed mice to a greater extent than NC-fed mice, as reported by cluster analysis at the family level (figure 1L).
We also investigated whether colonisation with periodontal pathogens may induce changes in the gut microbiota. In NC-fed mice, the above periodontal microbiota changes were associated with tiny modifications of gut microbiota due to colonisation, which increased members of Actinobacteria and Deltaproteobacteria groups (see online supplementary figure S2A). In HFD-fed mice, we also observed tiny modifications of gut microbiota due to colonisation (only unclassified bacteria), whereas HFD increased the intestinal proportion of Proteobacteria (see online supplementary figure S2B). All the gut microbial profiles in each group from Phyla to Species are provided in online supplementary figure S2C–H.
Altogether, this data set provides evidence that periodontal pathogens induce periodontitis in mice, alter periodontal microbiota and that the subsequent periodontitis aggravates HFD-induced periodontal and systemic inflammation.
Periodontitis increases HFD-induced metabolic diseases
To demonstrate that periodontal pathogen-induced periodontitis may represent an aggravating risk factor for diet-induced metabolic diseases, we characterised glucose metabolism in response to the nutritional stress. Our data show that periodontitis aggravated the HFD-induced glucose intolerance by the first and up to the third month of treatment (figure 2A, C, E). These data were associated with a progressive and significant increase in the fat/lean mass ratio (figure 2B, D, F). Furthermore, according to the time course, periodontitis increased plasma leptin and insulin concentrations on HFD at 3 months (table 1). Insulin resistance (indexed by the glucose-infusion rate (GIR)), as assessed by the euglycemic/hyperinsulinemic clamp technique, was induced by the periodontitis in HFD-fed mice only (figure 2G). Importantly, ABL was strongly and significantly correlated (R2=0.52; p<0.0001) with insulin resistance (figure 2H).
Then, we analysed by PCoA whether correlations may exist between families abundance in periodontal microbiota and metabolic parameters such as GIR, glycaemic index (GI3), fasted glucose (FG3), ABL, BW3, cytokines for gingival inflammation, blood immunoglobulins levels and gingival (IgG) cytokines at 3 months. We found that Lactobacillaceae abundance in periodontal microbiota positively and significantly correlated with GIR (figure 2I, up-left inset). By contrast, Porphyromonadaceae abundance was negatively and significantly correlated with GIR (figure 2I, up-right inset), but positively with ABL (figure 2I, down-right inset). Overall, the PCoA allowed identifying a specific cluster for each group, despite a tiny superposition between the NC-Co and the HFD group. Furthermore, Porphyromonadaceae abundance was associated with gingival TNF-α expression (figure 2I, down-left inset), whereas we also found a positive and significant correlation between ABL and fasted glycaemia at 3 months (figure 2I, middle-right inset).
Altogether, these data show that periodontitis aggravates HFD-induced glucose intolerance and insulin resistance together with changes in the periodontal microbiota, among which some of them are associated with the metabolic phenotypes.
Pg colonisation recruits cells from the adaptive immune system to control glucose tolerance
To demonstrate that the adaptive immune system was triggered by the change in periodontal microbial ecology and was a causal mechanism responsible for the deleterious impact of the periodontal pathogens on metabolic disease, we first transferred the cervical lymph-node cells from mice with or without periodontitis to healthy recipient mice (figure 3A). In such conditions, the glucose tolerance was similar in both groups of recipient mice (not shown), suggesting that other factors were required to trigger the metabolic disease. Hence, we challenged the recipient mice with the periodontal pathogens during 4 weeks after the cell transfer (figure 3A) and demonstrated that the glucose tolerance was improved in mice that received the immune cells from the infected mouse when compared to those that received immune cells from a non-infected mouse (figure 3B, C). BW gain was not significantly affected (not shown). Then, to study the impact of the colonisation and of the transfer of cervical immune cells from mice with periodontal disease on HFD-induced glucose intolerance, we then challenged the colonised and transferred mice for 4 weeks with HFD. After 4 weeks of HFD, the IPGTT is showing that the protection, although still significantly present in glycaemic index, is starting to vanish (figure 3D, E). The data show that when the HFD-fed mice were transferred with immune cells from mice with periodontitis, the glucose tolerance is improved although modestly (figure 3D, E). By contrast, the transfer of immune cells by itself, without colonising the recipient mice, was not sufficient to impact glucose tolerance (figure 3B–E). However, glucose metabolism could be impacted when the immune cells were specifically adapted to the periodontal pathogens. This suggests that both nutritional stress and periodontitis are required factors to trigger metabolic phenotypes.

Immune cells transfer from cervical lymph nodes from periodontitis mice reduce colonisation-induced glucose intolerance. (A) Immune cells from cervical lymph nodes from donor mice with or without periodontitis were transferred to recipient mice. Then, each group was colonised by Porphyromonas gingivalis (Pg), Fusobacterium nucleatum (Fn) and Prevotella intermedia (Pi) in periodontal tissue for 4 weeks. Intraperitoneal glucose-tolerance tests were performed in recipient mice after transfer (not shown), after colonisation (B) and after 4 weeks of high-fat diet (HFD) (D); (C) and (E) glycaemic index. After colonisation (±): HTC+NC-Co (black bar n=4): healthy transfer+colonisation, PTC+NC-Co (green bar n=4) periodontitis transfer+colonisation, HTC+NC (blue bar n=4) and PTC+NC (purple bar n=4) periodontitis transfer+no colonisation and after 4 weeks of HFD: HTC+HFD-Co (black bar n=4): healthy transfer+colonisation, PTC+HFD-Co (green bar n=4) periodontitis transfer+colonisation, HTC+HFD (blue bar n=4) and PTC+HFD (purple bar n=4) periodontitis transfer+no colonisation. Data are mean±SEM. Significant results when ***p<0.001 when compared to HTC+NC as determined by one-way analysis of variance (ANOVA) followed by Tukey's test (C) and two-way ANOVA with Bonferroni's post-test (B).
In a second set of experiments, to further validate the role of the adaptive immune system on the control of glucose tolerance, we immunised the lymphocytes to the periodontal pathogens by treating mice with different sets of inactivated periodontal pathogens (figure 4A). The intramuscular treatment with the three inactivated periodontal pathogens prevented the above reported aggravating effects of periodontitis on HFD-induced glucose intolerance at 1 month (see online supplementary figure S3A–F), 2 months (not shown) and 3 months (figure 4B–F). Importantly, this preventive effect was due to Pg since the treatment of the mice with this unique bacterium was sufficient to protect against periodontitis-induced metabolic diseases. Moreover, the specific treatment by Pg prevented the decrease of antibodies anti-Pg observed on HFD after periodontal colonisation at 1 and 3 months (see online supplementary figures S3G and S4G). At 3 months of HFD, the treatment with Pg protected against the periodontal colonisation-induced ABL in periodontal tissue (figure 4H).

Pre-treatment with inactivated Porphyromonas gingivalis (Pg) prevents periodontitis-aggravated glucose intolerance in high-fat diet (HFD)-fed mice. (A) Mice were injected by 106 colony-forming unit (CFU) of inactivated Pg or inactivated Fusobacterium nucleatum (Fn) or inactivated Prevotella intermedia (Pi) or a mix of all inactivated bacteria or vehicle solution. One month later, mice were colonised by Pg, Fn, Pi and/or by vehicle solution for 1 month and then randomised into seven groups: NC-Vehicles (vehicle+normal chow, black bar, n=4), HFD (vehicle+HFD, red bar, n=4), high-fat diet colonised (HFD-Co) (vehicle+HFD+colonisation, green bar, n=4), HFD-Co+I B mix (inactivated mix bacteria+colonisation+HFD, black blue bar, n=4), HFD-Co+I Pg (inactivated Pg+colonisation+HFD, purple bar, n=4), HFD-Co+I Fn (inactivated Fn+colonisation+HFD, light blue bar, n=4) and HFD-Co+I Pi (inactivated Pi+colonisation+HFD, orange bar, n=4). Intraperitoneal glucose-tolerance test (IpGTT) and glycaemic index were assessed for each group after 3 months of HFD (B–F); (G) measurement of immunoglobulin G antibodies specific to lipopolysaccharide (LPS) of Pg in blood. (H) Alveolar bone loss was explored after experimental procedures for each group. Data are mean±SEM. Significant results when: **p<0.01, ***p<0.001 and ****p<0.0001 when compared to HFD vehicles, §p<0.05 and §§§§p<0.0001 when compared to NC vehicles and #p<0.05 and ####p<0.0001 when compared to HFD-Co as determined by two-way analysis of variance (ANOVA) with Bonferroni's post-test for (B), (C), (D) and (E) and one-way ANOVA followed by Tukey's post-test for (F) and (G).
The LPS from Pg enhance HFD-induced metabolic diseases
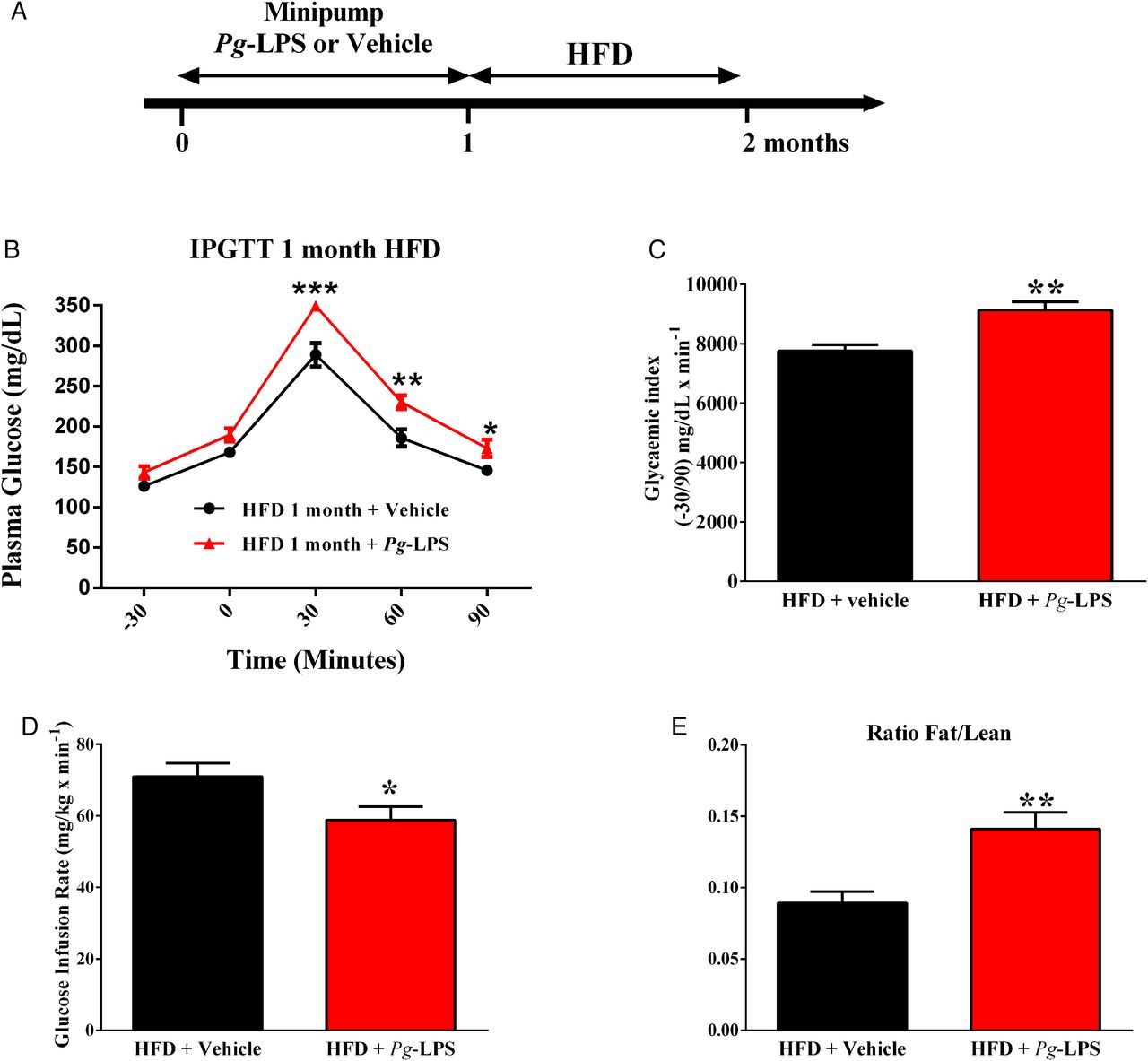
In the quest for molecular determinants responsible for Pg-aggravated diet-induced metabolic diseases, we evaluated the impact of LPS from Pg (figure 5A). A 1-month continuous infusion of a low rate of Pg-LPS aggravated HFD-induced glucose intolerance and insulin resistance (figure 5B–D). Furthermore, the fat to lean ratio was significantly increased in mice pre-treated with Pg-LPS when compared to saline (vehicle)-pre-treated mice (figure 5E), suggesting that the LPS from Pg could be a major molecular mechanism explaining the metabolic impact of periodontitis on metabolic disease.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lipopolysaccharide (LPS) from Porphyromonas gingivalis (Pg) aggravates high-fat diet (HFD)-induced metabolic diseases. (A) Mice were implanted with Pg-LPS or vehicle releasing minipumps for 1 month; then, mice were fed an HFD for 1 month. After 1 month of diet, glucose-tolerance (B), glycaemic index (C), insulin sensitivity (E) and ratio fat/lean mass (D) were assessed in each group: (Pg-LPS, red bar, n=7) or (NaCl, black bar, n=7). Significant results when: *p<0.05, **p<0.01 and ***p<0.001 when compared to HFD+vehicle as determined by one-way analysis of variance (ANOVA) followed by Tukey's test, except for ‘B’ (two-way ANOVA and Bonferroni's post-test).
Discussion
Here, we report that periodontitis and notably the LPS from Pg are aggravating factors for HFD-induced glucose intolerance and insulin resistance. The underlying mechanism is associated with the capacity to produce specific antibodies against Pg.
By using our unique animal model, we show that, in mice at risk of developing metabolic diseases (ie, HFD-fed), the prior colonisation of periodontal tissue with Pg enhances the development of glucose intolerance by affecting the adaptive immune system response against Pg. This set of data confirms evidences in humans where the incidence of metabolic diseases is further raised when associated with periodontitis,2 ,3 an epidemiological feature also observed in pre-diabetic or undiagnosed T2D patients.4 ,5 Our data show that the periodontitis itself was not sufficient to induce metabolic impairments since no change in glycaemia or in insulin resistance was observed in infected NC-fed mice. The corroborating factor brought by HFD could be linked to the gut microbiota dysbiosis characterised in patients with impaired metabolism.14 However, periodontitis appeared to induce no major changes in the gut microbiota of colonised mice. Conversely, periodontitis induced a profound periodontal microbiota dysbiosis, aggravated on HFD.
Among periodontal pathogens, Pg is a key candidate in the relationship between periodontitis and general health.32 Pg has a specific arsenal of virulence factors that enable it to invade the periodontal tissue and subsequently disseminate into the systemic circulation. First, the ability of Pg to invade the periodontal biofilm is regulated by specific enzymes such as the bacterial dipeptidyl peptidase 4 (DPP4).33 As an indication, inhibitors of DPP4 are currently used to treat T2D, suggesting that the efficacy of this therapeutic strategy may include the inhibition of prokaryotic enzymes.34 Indeed, Pg is also capable of invading host cells such as epithelial and endothelial cells.35 Pg is captured by host cells through interactions between its fimbriae and α5β1-integrin expressed by the epithelial cells, allowing it to cross the epithelial barrier.36 Altogether, these observations confer an important role of Pg on its dissemination. Here, we show that part of Pg virulence specifically attributed to metabolic disease is through LPS since the continuous low rate infusion of LPSs from Pg, together with an HFD, impacted on the immune system and consequently on insulin and glucose tolerance. By providing a mechanism, our conclusions thus confirm and further extend the data from the literature, which show that patients with periodontitis are characterised by the presence of Pg-LPS into the blood.32 The continuous infusion of LPS from Pg may induce a chronic low-grade systemic inflammation leading to the resetting of cardiac homeostasis.37 Such a mechanism involves the toll-like receptors (TLRs) TLR-2 and TLR-438 leading to the activation of MyD88 and nuclear factor κB (NF-κB) responsible for the transcription of pro-inflammatory cytokines genes such as TNF-α or IL-1β.38 Furthermore, Pg LPS-stimulated gingival fibroblast showed NF-κB-dependent TNF-α production leading to an inflammatory process involving ERK, p38 and JNK activation, as well as PAI-1 expression.39 This mechanism was also observed in stem cells from human periodontal ligament.40 The role of LPS to cellular cross-talk is essential to the virulence of Pg. We previously provided a mechanism by showing that mice deprived of the LPS receptor CD14 did not develop metabolic10 ,41 and periodontal22 diseases, which reinforces our present conclusion. We cannot rule out that the putative role of periodontitis on metabolic disease could be linked to changes of gut microbiota. The sequencing data showed some subtle changes following the colonisation in NC-fed mice. However, no impact on glucose nor on immune homeostasis was detected in our experimental conditions. Therefore, it could be expected that the periodontitis could slowly impair metabolic features over a longer period of time even in the absence of nutritional stress. But we cannot rule out that such mechanisms, as reported elsewhere,42 could have some long-term impact in the present experimental conditions. The subtle changes in gut microbiota reported here have most likely no influence on glucose homeostasis. The mechanism through which Pg would favour the development of metabolic diseases could be due to the strong impact of microbiota dysbiosis on the immune response.43 We show that following the action of Pg, a pro-inflammatory process is triggered. The nutritional stress impacted the cervical and systemic immune response mostly from the innate immune cells with a mild impact on B-lymphocyte and T-lymphocyte. However, when combined with the nutritional stress (HFD), this response was normalised, suggesting that the low-grade inflammation induced by the innate immune system was under the control of the adaptive immune system. We demonstrated this hypothesis by showing that the transfer of cells from the cervical lymph nodes of infected mice to naive recipient mice prevented a further impact of periodontitis on metabolic parameters. Therefore, the lymphocytes from the colonised mice were educated to the periodontal antigens and protected the recipient mouse from the metabolic inflammation. Furthermore, we could notice the production of the antibodies anti-Pg in response to the treatment with the corresponding attenuated bacteria. The production of the antibody anti-Pg was associated with the prevention of the development of metabolic disease, suggesting the protective role of the antibodies, at least in part. The role of the innate and adaptive immune system and notably the role of the molecule TREM-1 have been suggested44 and in response to the LPS from Pg as assessed by the enhancement of IL-1β and IL-6 secretion.45 There is increasing evidence for the role of B, T and natural killer lymphocytes in the development of DT2.46 Pg increases the presence of IL-17 and Th17 cells in human periodontitis lesions.47 This has also been observed in arthritis and obesity promoted by periodontitis.48 ,49 Importantly, the presence of bacteria at the site of infection could lead to a systemic inflammation in case of an impaired immune system.50 This hypothesis from the literature is supported by our data showing that the systemic immune system was dramatically affected by the periodontal infection. Our data showed that the vaccination procedure using Fn or Pi alone could not protect against periodontitis-induced metabolic alteration. This suggests that without Pg the former pathogens are not sufficient. However, we cannot rule out that Fn and Pi could be worsening factors for Pg virulence.
Finally, altogether our data show that a periodontal microbiota dysbiosis induced upon a fat-enriched diet impacts on cervical lymph nodes and systemic immune system response. Although the nutritional stress and the gut microbiota dysbiosis can induce metabolic diseases per se, here we further show that a concomitant periodontal disease triggers the regional adaptive immune system, which enhances insulin resistance. Our data support a role for molecular factors issued from Pg, such as LPS. The latter is considered as an adjuvant for the periodontitis induced by the nutritional stress, which altogether trigger the adaptive immune response to enhance both insulin resistance and glucose intolerance.
Acknowledgments
The authors thank the French Society of Arterial Hypertension (Société Française d'HyperTension Artérielle) and the French Diabetes Society (Société Francophone du Diabète) for supporting Dr Blasco-Baque. They also thank the zootechnie-Rangueil INSERM/UPS US006 CREFRE; the Phenotypage-ANEXPLO Platform (US006-CREFRE) for biochemical assays; the microtomography facility in the medical faculty of the University Jean Monnet (St. Etienne, France) and especially Dr Luc Malaval (INSERM U1059) for technical support; Florence Capilla and Christine Salon for technical assistance in histology for hemi-mandibles at the Platform of Experimental Histopathology of the INSERM/UPS US006 CREFRE, Toulouse Purpan, France; Catherine Le Iann for technical assistance in P. gingivalis antibodies measurement; Lorette Gaffié and Dr Robert Cameron for editing the English and Dr Tercé François for critical reviewing of the manuscript. The authors also thank technical Platform of Research in Odontology from University of Paul Sabatier Toulouse.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors VB-B performed and designed experiments, analysed data, wrote and revised the manuscript. LG, CP, QE, SN, PL, SLG-D, ML, PK, AW, VA, AC and MB-M performed experiments. MS performed experiments, analysed data, wrote and revised the manuscript. RB designed experiments, analysed data and wrote and revised the manuscript. All authors have approved the final version to be published.
Competing interests None declared.
Ethics approval All animal experimental procedures were approved by the local ethical committee of Rangueil University Hospital (Toulouse, France).
Provenance and peer review Not commissioned; externally peer reviewed.