Article Text

Abstract
Objective α-Conotoxin Vc1.1 is a small disulfide-bonded peptide from the venom of the marine cone snail Conus victoriae. Vc1.1 has antinociceptive actions in animal models of neuropathic pain, but its applicability to inhibiting human dorsal root ganglion (DRG) neuroexcitability and reducing chronic visceral pain (CVP) is unknown.
Design We determined the inhibitory actions of Vc1.1 on human DRG neurons and on mouse colonic sensory afferents in healthy and chronic visceral hypersensitivity (CVH) states. In mice, visceral nociception was assessed by neuronal activation within the spinal cord in response to noxious colorectal distension (CRD). Quantitative-reverse-transcription-PCR, single-cell-reverse-transcription-PCR and immunohistochemistry determined γ-aminobutyric acid receptor B (GABABR) and voltage-gated calcium channel (CaV2.2, CaV2.3) expression in human and mouse DRG neurons.
Results Vc1.1 reduced the excitability of human DRG neurons, whereas a synthetic Vc1.1 analogue that is inactive at GABABR did not. Human DRG neurons expressed GABABR and its downstream effector channels CaV2.2 and CaV2.3. Mouse colonic DRG neurons exhibited high GABABR, CaV2.2 and CaV2.3 expression, with upregulation of the CaV2.2 exon-37a variant during CVH. Vc1.1 inhibited mouse colonic afferents ex vivo and nociceptive signalling of noxious CRD into the spinal cord in vivo, with greatest efficacy observed during CVH. A selective GABABR antagonist prevented Vc1.1-induced inhibition, whereas blocking both CaV2.2 and CaV2.3 caused inhibition comparable with Vc1.1 alone.
Conclusions Vc1.1-mediated activation of GABABR is a novel mechanism for reducing the excitability of human DRG neurons. Vc1.1-induced activation of GABABR on the peripheral endings of colonic afferents reduces nociceptive signalling. The enhanced antinociceptive actions of Vc1.1 during CVH suggest it is a novel candidate for the treatment for CVP.
- ABDOMINAL PAIN
- VISCERAL NOCICEPTION
- IRRITABLE BOWEL SYNDROME
- ION CHANNELS
- REAL TIME PCR
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Patients with IBS suffer from chronic visceral pain (CVP); however, there are limited analgesic therapeutic options currently available for treatment.
A rich source of novel agents to treat chronic pain is the α-conotoxin family of peptides from the venom of marine cone snails.
α-Conotoxin Vc1.1 has antinociceptive and antihyperalgesic actions in neuropathic pain models; however, its ability to inhibit human sensory dorsal root ganglion (DRG) neurons remains unknown.
Vc1.1's applicability in reducing CVP is also unknown.
What are the new findings?
Vc1.1 reduces human sensory DRG neuroexcitability, via a γ-aminobutyric acid receptor B (GABABR)-mediated mechanism.
We show that human DRG neurons express GABABR and the voltage-gated calcium channels CaV2.2, and CaV2.3, which are the direct and downstream targets of Vc1.1, respectively.
Vc1.1 inhibits mouse colonic nociceptors and also low-threshold distension-sensitive colonic afferents with greatest effect during chronic visceral hypersensitivity (CVH).
Peripheral in vivo Vc1.1 administration inhibits the signalling of noxious information from the colon to the spinal cord. This antinociceptive effect is also greater in mice with CVH.
During CVH, mouse colonic DRG neurons show upregulation of the CaV2.2 exon-37a variant, which may explain the increased inhibitory effect of Vc1.1 in CVH states.
How might it impact on clinical practice in the foreseeable future?
Vc1.1 has been tested in human clinical trials for the treatment of neuropathic pain, where it has been demonstrated to have a clean safety and side-effect profile.
Our current findings show Vc1.1 inhibits human DRG neurons, via activation of the GABABR, which is a key finding for clinical translatability.
This inhibitory effect in human neurons combined with the enhanced antinociceptive action of Vc1.1 in colonic pathways in a mouse model of CVH suggests it is a novel candidate for the treatment for CVP associated with IBS.
We show that by reducing nociceptive signalling from the periphery, Vc1.1 has potential therapeutic value in treating CVP.
These findings put GABABR agonists in the spotlight as potential peripheral agents for the treatment of CVP.
Introduction
IBS is a prevalent, chronic GI disorder that negatively impacts the quality of life in up to 14% of the population.1 ,2 It is characterised by abdominal pain and discomfort associated with altered bowel habits.3–5 Although the pathophysiology of IBS is not completely understood, it is becoming clear that changes to peripheral cellular and sensory mechanisms play key roles in the associated pain.6 ,7 In particular, chronic visceral hypersensitivity (CVH) of colonic afferents is implicated in the development and maintenance of chronic visceral pain (CVP) in IBS.4 ,5 Characteristic features of CVH include nociceptor hypersensitivity8 and increased signalling of noxious colorectal distension (CRD) within the spinal cord.9–11 Recent evidence suggests sensory afferents display upregulation of numerous ion channels and receptors in animal models of CVH,7 ,10 ,12 making them targets for analgesic treatment.
A recently introduced treatment for patients with IBS and constipation involves a small disulfide-rich peptide that is restricted to the GI tract, where it inhibits peripheral nociceptive pathways and produces clinically relevant pain relief.9 Given the limited treatments available for patients with other subtypes of IBS, additional analgesic therapeutic options are needed. A rich source of novel small disulfide-rich agents comes from the α-conotoxin family of peptides from the venom of marine cone snails.13 These peptides target a wide variety of membrane receptors and ion channels.14 In particular α-conotoxin Vc1.1, a 16-amino acid synthetic version of a peptide derived from the marine cone snail Conus victoriae, has antinociceptive actions in vitro and antihyperalgesic actions in numerous in vivo neuropathic pain models.15–17 Interestingly, in a chronic constriction injury model of neuropathic pain, Vc1.1 relieves tactile allodynia.17 These inhibitory effects were similar to those obtained with gabapentin, a ligand recently proposed as a potential IBS therapeutic,18 but were achieved at far lower doses.17 Notably, Vc1.1 (also called ACV1) has been used in phase I and phase IIA clinical trials for the treatment of neuropathic pain.19–21 These studies showed Vc1.1 was safe and well tolerated with a clean safety and side-effect profile. Despite this promise, therapeutic trials were discontinued as Vc1.1 was shown to be less potent at the human α9α10 nicotinic acetylcholine receptor (nAChR), which was thought to mediate the inhibitory action of Vc1.1. However, more recent recombinant cell line studies have clearly demonstrated that the human γ-aminobutyric acid receptor B (GABABR) is the primary and high affinity target for Vc1.1.17 ,22 ,25–27 These studies also demonstrated GABABR activation by Vc1.1 causes downstream inhibition of the voltage-gated calcium channels CaV2.2 and CaV2.3, which underlies Vc1.1's inhibitory actions.14 ,28 These recent findings are intriguing; as both oral and intravenous administration of baclofen, the archetypal GABABR agonist has been shown to reduce the pseudo-affective responses to CRD in animal models.29 ,30 Although it is unclear if this baclofen-induced inhibition is centrally or peripherally mediated, we wondered if Vc1.1 represents a novel peripheral gut analgesic for the treatment of CVP. Therefore, we determined if Vc1.1 inhibits human sensory dorsal root ganglion (DRG) neurons, the primary transducers at the start of the pain-processing pathway. Second, we determined if Vc1.1 inhibits sensory pathways within the splanchnic and pelvic innervation of the colon and whether these actions are enhanced in an animal model of CVH. Third, we determined if the inhibitory actions of Vc1.1 are mediated via activation of GABABR on the peripheral endings of colonic afferents.
Materials and methods
For comprehensive descriptions of the methodologies used, see the online supplementary information.
Human DRG
Thoracolumbar (TL) DRG (T9–L1) were acquired from five (three female, two male) human adult organ donors (22.2±2.08 years of age) during the removal of the vital organs for transplantation. The harvested DRG were immediately processed for downstream patch clamp or RNA studies. Intact DRG were kept for quantitative-reverse-transcription-PCR (qRT-PCR) mRNA expression studies from each spinal level (T9, T10, T11, T12, L1) while additional DRG were dissociated to allow individual DRG neurons to be studied with single-cell-reverse-transcription-PCR (RT-PCR) studies, or to allow patch clamp recordings to be performed.
Human DRG patch clamp recordings
Whole-cell patch clamp recordings of cultured human DRG neurons were performed in current clamp mode in response to depolarising current pulses (20 or 50 pA current steps, 500 ms duration). This allowed the rheobase (amount of current needed to initiate action potential generation) to be assessed in the presence and absence of Vc1.1 (1000 nM) and a synthetic analogue of Vc1.1 ([P6O]Vc1.1;1000 nM), which is inactive at GABABR. An increased rheobase indicates more current is required to fire an action potential and therefore the neuron displays reduced excitability.
Mouse model of CVH
Intracolonic trinitrobenzene-sulfonic acid (TNBS) was administered as described previously.8–10 TNBS-treated mice were allowed to recover for 28 days, at which stage inflammation had resolved and chronic colonic afferent mechanical hypersensitivity was evident.8–10 ,12
Ex vivo electrophysiology
Recordings of splanchnic and pelvic afferents were made from healthy control and CVH mice as described previously.8–10 Briefly, colonic nociceptors were recorded from the splanchnic pathway. They respond to noxious distension (40 mm Hg), stretch (≥7 g) or von Frey hair filaments (2 g)8 ,31 and become mechanically hypersensitive in models of CVP.8–10 ,12 Muscular–mucosal afferents were recorded from the pelvic pathway and respond to both low-intensity circular stretch (<5 g) and fine mucosal stroking (10 mg).8 ,31–33 Once baseline responses had been established, mechanosensitivity was retested after application of Vc1.1 (1, 10, 100, 1000 nM) for 10 min at each dose. To determine the mechanism of action of Vc1.1 the selective GABABR antagonist (CGP55845:5 μM), CaV2.2 blocker (ω-conotoxin CVID:1 μM) or CaV2.3 blocker (SNX-482:200 nM) were applied alone, or in combination, at maximally effective concentrations for 10 min prior to coincubation with Vc1.1 (1000 nM).
CRD and pERK immunohistochemistry
Healthy control or CVH mice received an intracolonic enema of either saline or Vc1.1 (1000 nM). Ten minutes later, under anaesthesia, a 4 cm CRD balloon catheter was inserted transanally into healthy or CVH mice.9–11 After regaining consciousness CRD was performed (80 mmHg for 10 s, deflated for 5 s, repeated five times). Following sacrifice via anaesthetic overdose, mice underwent fixation by transcardial perfusion and the TL (T10–L1) and lumbosacral (LS:L6–S1) spinal cord removed and cryoprotected. Frozen sections were cut and incubated with monoclonal rabbit anti-phosphorylated MAP kinase ERK 1/2 (pERK) and AlexaFluor-488 was used for visualisation.9–11
Isolation of mouse colonic DRG neurons
TL and LS DRG were removed from healthy control and CVH mice 4 days after retrograde tracing from the colon with AlexaFluor-555-conjugated cholera-toxin subunit-B (CTB-AF555). DRG were dissociated and single or pooled colonic DRG neurons isolated for downstream mRNA expression analysis.10
Quantitative-reverse-transcription-PCR
RNA was extracted from either whole human DRG and single human DRG neurons or mouse whole DRG, pooled colonic DRG neurons and single colonic DRG neurons from healthy control and CVH mice using specific isolation kits. QRT-PCR was performed using either human-specific or mouse-specific primers for GABABR1, GABABR2, CaV2.2 and CaV2.3.10 ,12 ,32 ,34 The comparative cycle threshold method was used to quantify the abundance of target transcripts to reference genes.10 ,12 ,32 ,34
Immunohistochemistry
In both perfused-fixed frozen mouse DRG sections and dissociated mouse DRG neurons specific antibodies for GABABR1, GABABR2, CaV2.2 or CaV2.3 were used to determine the expression of these targets in retrogradely traced colonic DRG neurons. Antibody preabsorption and omission of primary antibodies were used as controls (see online supplementary figure S1). AlexaFluor-488 or AlexaFluor-594 conjugated secondary antibodies were used for visualisation.
Results
Vc1.1 reduces the excitability of human DRG neurons
To determine if Vc1.1 reduces the excitability of human DRG neurons we used whole-cell patch clamp recordings to assess neuronal excitability. Vc1.1 (1000 nM) inhibited a specific population (40%) of human DRG neurons, which was indicated by a significant increase in the amount of injected current required to fire an action potential (figure 1Ai). In this population of neurons, Vc1.1 increased the rheobase by 20% compared with control responses (figure 1Aii, B and see online supplementary figure S2). The average of cell capacitance for all the recorded human DRG neurons was 131.48±18.03 pF, with no significant difference in cell capacitance observed between neurons which were affected by Vc1.1 and those that were not.
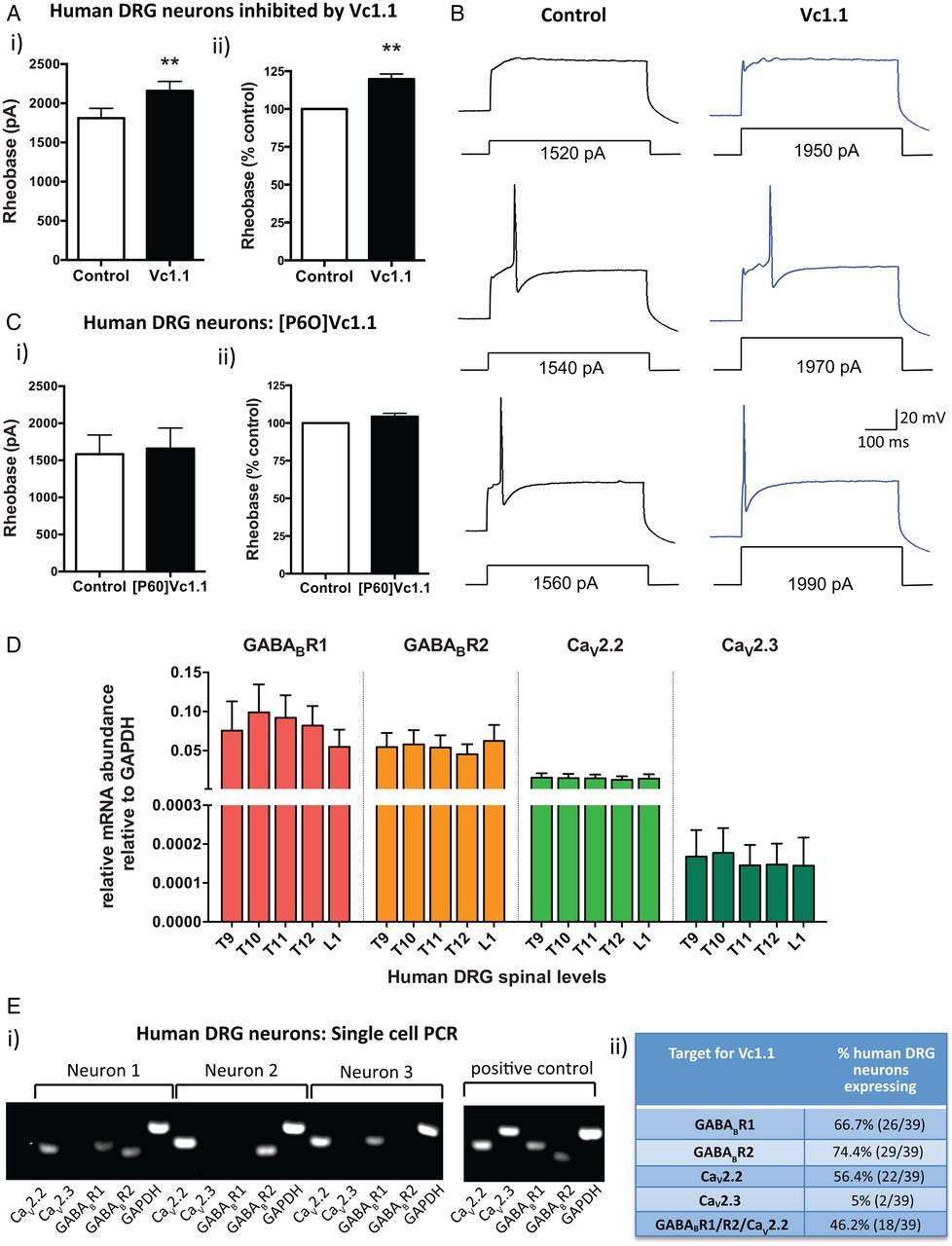
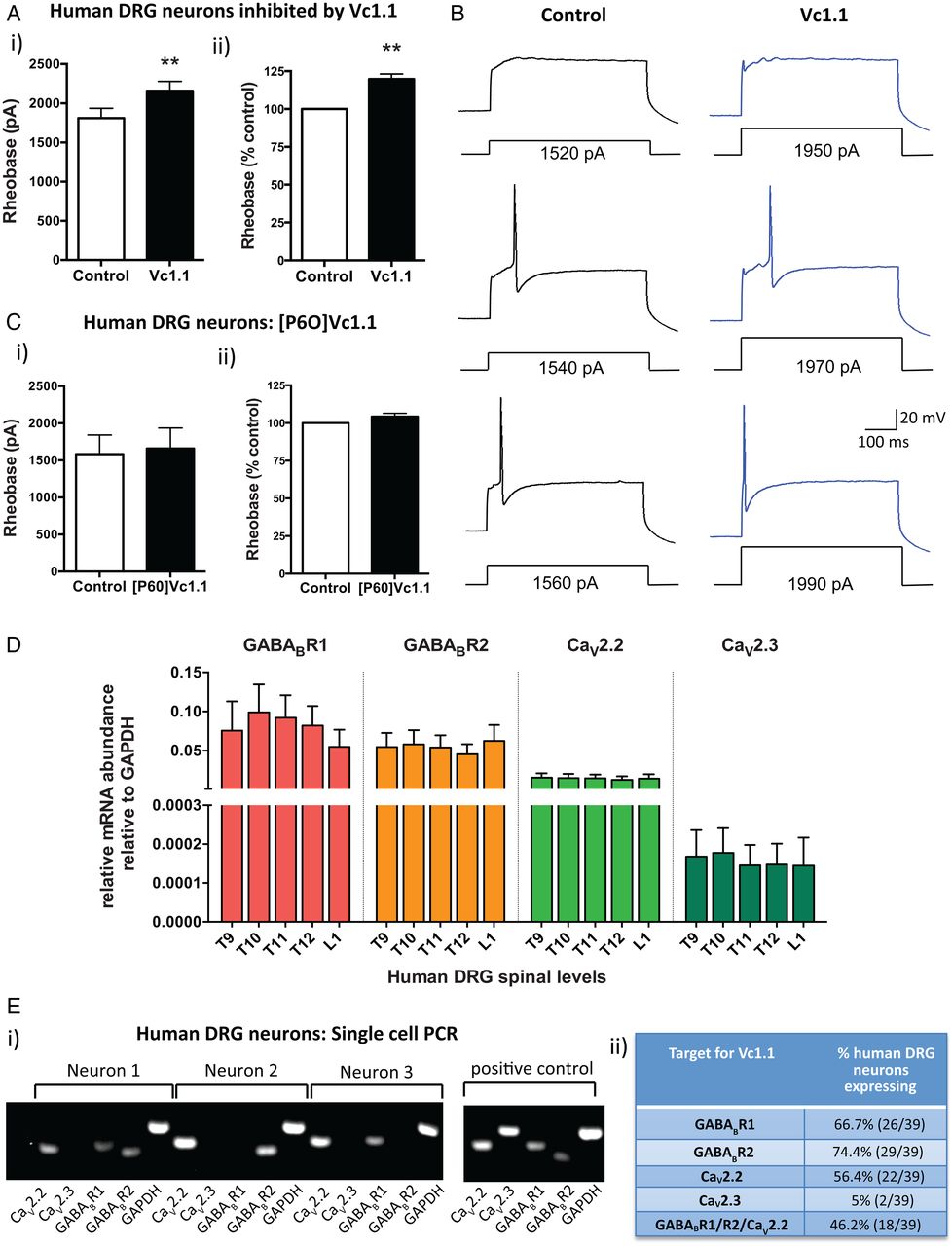
α-Conotoxin Vc1.1 inhibits human dorsal root ganglion (DRG) neurons. (A) (i) Group data showing that Vc1.1 (1000 nM) significantly increases the rheobase of a subpopulation (40%) of human DRG neurons, indicating Vc1.1 inhibits neuronal excitability and more current is required to initiate an action potential (**p<0.001, n=10, paired t-test). (ii) In this population of neurons, Vc1.1 increased the rheobase by 20% compared with baseline response, meaning the neurons are less excitable (**p<0.001). (B) Representative examples of human DRG neuronal responses in the absence (control solutions) and in the presence of Vc1.1. Note in this example more current is required to fire an action potential from a human DRG neuron in the presence Vc1.1 (1970 pA) relative to control (1540 pA). (C) [P6O]Vc1.1 (1000 nM), a synthetic analogue of Vc1.1 that does not act at γ-aminobutyric acid receptor B (GABABR), did not affect human DRG neuronal excitability when expressed as either i) rheobase or ii) % of rheobase (p>0.05, n=10, paired t-test) indicating Vc1.1 induces its inhibitory effect via the GABABR. (D) Group data of quantitative-reverse-transcription-PCR analysis from thoracolumbar (T9, T10, T11, T12, L1) DRG from four human adult donors indicating expression of the GABABR subunits R1, R2 and the voltage-gated calcium channels CaV2.2 and CaV2.3 in human DRG. (E) (i) Examples of gel electrophoresis following single-cell-PCR analysis from individual human DRG neurons. (ii) Combined analysis of expression and coexpression of GABABR and CaV channels in 39 human DRG neurons. Of human DRG neurons, 46.2% (18/39) coexpress GABABR and CaV2.2, the minimum components required for Vc1.1-induced inhibition. Combined these studies indicate Vc1.1 inhibits human DRG neurons via a GABABR-mediated mechanism. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To determine if this inhibition was mediated via GABABR or α9α10-nAChR we used a modified version of Vc1.1 ([P6O]Vc1.1), which is inactive at the GABABR, but active at the α9α10-nAChR.35 [P6O]Vc1.1 had no effect on human DRG neuronal excitability (figure 1C), suggesting Vc1.1 exerts its inhibitory effects on human DRG neurons via a GABABR mechanism. Recent recombinant cell line studies have demonstrated that the human GABABR is the high affinity (nanomolar) target for Vc1.1 and that GABABR activation by Vc1.1 causes downstream inhibition of the voltage-gated calcium channels CaV2.2 and CaV2.3.14 In order to determine if the same mechanism applied in human DRG neurons, we determined the expression of GABABR and CaV channels in whole TL DRG from five spinal levels from four human adult donors. We showed that subunits R1 and R2 for GABABR were expressed as well as CaV2.2 and CaV2.3 (figure 1D). Expression levels for each of the targets were remarkably consistent between the different DRG levels across the four human donors (figure 1D). Single-cell-RT-PCR of individual human TL DRG neurons demonstrated that 46% coexpressed GABABR and CaV2.2, the minimum components required for Vc1.1-induced inhibition (figure 1E). This was consistent with our patch clamp observations where 40% of the human DRG neurons tested were inhibited by Vc1.1. Overall, these functional and expression studies indicate Vc1.1 inhibits human DRG neurons via a GABABR-mediated mechanism.
Vc1.1 inhibits mouse colonic afferents with greater efficacy in CVH
Given Vc1.1's inhibitory actions on human DRG neurons in the current study and rat somatosensory neurons in previous studies,16 ,35 ,36 we hypothesised Vc1.1 may also inhibit colonic afferents. To test this hypothesis we performed in vitro single-unit colonic afferent recordings.9 ,10 ,32 ,33 Specifically, we recorded from mouse splanchnic nerves, which supply the mid-to-distal colon and signal predominantly nociceptive information,8 ,33 and the pelvic nerves supplying the colorectum, which signal a mixture of physiological and nociceptive information.8 ,33 Vc1.1 significantly and dose-dependently decreased healthy colonic nociceptor activity, with a maximum reduction in response to mechanical stimulation of 32% at the highest concentration tested (figure 2Ai). We then asked if these Vc1.1-induced antinociceptive effects were maintained, or indeed augmented, in CVH. This question was assessed in a mouse model where colonic nociceptor mechanical hypersensitivity7–10 and colonic mechanical hyperalgesia and allodynia are evident long after resolution of TNBS-induced colitis.7 ,37 ,38 We found that colonic nociceptors in the CVH model displayed pronounced hypersensitivity and that Vc1.1 significantly reduced nociceptor mechanosensitivity, showing significant reductions at 100 nM and 1000 nM, with a maximal reduction of 44% at 1000 nM (figure 2Aii). Overall, Vc1.1's inhibitory effect was greatly enhanced in CVH nociceptors compared with healthy nociceptors (figure 2B, C). We also tested whether the inhibitory effects of Vc1.1 extended to low-threshold distension sensitive pelvic afferents and found that Vc1.1 dose-dependently inhibited pelvic muscular–mucosal afferent responses to circular stretch in healthy mice (figure 2Di, Fi). The inhibitory effect of Vc1.1 on pelvic afferents was also enhanced in afferents from CVH mice (figure 2Dii, E, F).

α-Conotoxin Vc1.1 inhibits colonic afferents in ex vivo recordings from healthy and chronic visceral hypersensitivity (CVH) mice. (A) (i) Vc1.1 significantly inhibits splanchnic colonic nociceptors from healthy mice. Compared with baseline, Vc1.1 at 1000 nM significantly reduced colonic nociceptor mechanosensitivity (**p<0.01, n=10 afferents, one-way ANOVA, Bonferroni post hoc). (ii) In a model of CVH, nociceptors are potently and concentration-dependently inhibited by Vc1.1, with significant reductions in mechanical responses at 100 nM and 1000 nM (**p<0.01, n=10 afferents, one-way ANOVA, Bonferroni post hoc tests). (B) Change in mechanosensitivity induced by Vc1.1 in healthy and CVH nociceptors compared with their respective baseline responses. Vc1.1 caused significantly more inhibition at 100 nM (**p<0.01) and 1000 nM (*p<0.05) in CVH nociceptors than healthy nociceptors (healthy: n=10; CVH: n=10, two-way ANOVA, Bonferroni post hoc). (C) Single-unit recordings from the splanchnic innervation showing inhibition of (i) a healthy nociceptor, and (ii) a CVH nociceptor following application of Vc1.1 (1000 nM). (D) (i) Vc1.1 inhibited pelvic low-threshold muscular-mucosal afferents from healthy mice. Compared with baseline, significant reductions in the mechanosensory response evoked by 5 g circular stretch were observed at 100 nM and 1000 nM Vc1.1 (*p<0.05, n=6 afferents). (ii) Low-threshold muscular–mucosal afferents from CVH mice were also concentration-dependently inhibited by Vc1.1, with significant reductions at 100 nM (*p<0.05) and 1000 nM (**p<0.01, n=6 afferents). (E) Compared with their respective baseline responses, Vc1.1 causes significantly more inhibition of muscular–mucosal afferents (*p<0.05 at 1000 nM) from CVH mice (n=6) relative to healthy (n=6) mice. (F) Single-unit recordings from the pelvic innervation showing inhibition of (i) a healthy low-threshold muscular–mucosal afferent and (ii) a CVH muscular–mucosal afferent following application of Vc1.1 (1000 nM).
Vc1.1 reduces in vivo processing of noxious CRD in the mouse TL and LS spinal cord
Vc1.1 inhibits mouse colonic nociceptors in the splanchnic pathway and low-threshold distension sensitive afferents in the pelvic pathway. We therefore hypothesised these actions should correspondingly reduce signalling of noxious CRD relayed into the TL and LS spinal cord in vivo. In response to noxious CRD, pERK-immunoreactivity (pERK-IR) identifies activated neurons in the dorsal horn (DH) of the spinal cord.9–11 In healthy mice given noxious CRD, prior intracolonic administration of 1000 nM Vc1.1 resulted in significantly fewer pERK-IR DH neurons in both the TL and LS spinal cord (figure 3A–C).

Intracolonic administration of Vc1.1 reduces nociceptive signalling in the dorsal horn (DH) of the spinal cord in response to noxious colorectal distension (CRD). (A) Noxious CRD (80 mm Hg) in healthy mice results in activation of DH neurons in the thoracolumbar (T10–L1; splanchnic innervation) and lumbosacral (L6–S1; pelvic innervation) spinal cord, as indicated by pERK-immunoreactivity (pERK-IR). Mice pretreated with intracolonic Vc1.1 (1000 nM) display significantly fewer DH neurons in the thoracolumbar spinal cord, specifically T11–T12 (**p<0.01) and T13–L1 (**p<0.01), and the lumbosacral spinal cord (***p<0.001, one-way ANOVA, n=6: healthy+saline, n=6: healthy+1000 nM Vc1.1). (B) Schematic representation of laminae I–V (LI–LV) in the DH of the spinal cord. (C) (i) Healthy thoracolumbar DH. Left panel: following noxious CRD, pERK-IR (yellow arrows) neurons were predominantly located in the superficial DH (laminae I) and laminae V. Right panel: in healthy mice pretreated with Vc1.1 (1000 nM) fewer pERK-IR neurons are evident following noxious CRD. (ii) Healthy lumbosacral DH. Left panel: following noxious CRD, pERK-IR (yellow arrows) neurons were located in laminae I, II, IV and V. Right panel: healthy mice pretreated with Vc1.1 (1000 nM) displayed fewer pERK-IR neurons following noxious CRD, particularly within laminae I. (D) In chronic visceral hypersensitivity (CVH) mice, more neurons are activated by noxious CRD at baseline in the thoracolumbar DH. Pretreatment with intracolonic Vc1.1 (1000 nM) significantly reduces the number of pERK-IR DH neurons within the T10–T11(*p<0.05), T11–T12 (**p<0.01), T13–L1(**p<0.01) and lumbosacral DH (**p<0.01; CVH+intracolonic saline: n=6, CVH+intracolonic Vc1.1: n=6). (E) (i) Left panel: in CVH mice, following noxious CRD, pERK-IR neurons in the thoracolumbar DH were predominantly located in laminae I–II and throughout laminae III–V. Right panel: CVH mice pretreated with Vc1.1 (1000 nM) display fewer pERK-IR neurons following noxious CRD, particularly in the superficial laminae. (ii) CVH mice pretreated with intracolonic Vc1.1 (1000 nM) display fewer pERK-IR neurons in the lumbosacral DH.
In response to noxious CRD, CVH mice displayed greater numbers of pERK-IR DH neurons than healthy mice, which corresponds with the mechanical hypersensitivity of colonic nociceptors observed in our afferent recording studies. CVH mice pretreated with intracolonic Vc1.1 displayed significantly reduced numbers of pERK-IR DH neurons in both the TL and LS spinal cord following noxious CRD (figure 3D,E), with the extent of inhibition greater within the TL pathway (figure 3D). Overall, these results suggest Vc1.1 reduces the signalling of noxious stimuli from the colon and reduces the CVH observed in vivo.
Vc1.1-induced inhibition of mouse colonic afferents is meditated via the GABABR
To elucidate the site and mechanism of action of Vc1.1 in colonic pathways, we first used [P6O]Vc1.1, which is inactive at GABABR. As per our recordings in human DRG neurons, [P6O]Vc1.1 did not inhibit mouse colonic nociceptors (figure 4A), suggesting that the inhibitory effects of Vc1.1 on colonic afferents are mediated via GABABR.

The inhibitory effect of Vc1.1 on mouse colonic afferents is mediated via the γ-aminobutyric acid receptor B (GABABR). (A) The modified peptide (P6O)Vc1.1, which is inactive at the GABABR, does not inhibit colonic nociceptors from chronic visceral hypersensitivity (CVH) mice (Not Significant (NS), n=6 afferents, paired t-test). (B) (i) Application of the GABABR agonist baclofen caused dose-dependent inhibition of healthy colonic nociceptors, with significant reductions in mechanosensitivity observed at 20 μM (*p<0.05) and 200 μM baclofen (***p<0.001), respectively. (ii) Similarly, in colonic nociceptors from CVH mice, baclofen caused significant inhibition at 2 μM (*p<0.05), 20 μM (***p<0.001) and 200 μM (***p<0.001), respectively. (C) Change in mechanosensitivity induced by baclofen in healthy and CVH nociceptors, compared with their respective baseline responses. Baclofen caused significantly more inhibition at 20 μM (*p<0.05) and 200 μM (**p<0.01) in CVH nociceptors compared with healthy nociceptors (healthy: n=7; CVH: n=6, two-way ANOVA, Bonferroni post hoc). (D) (i) A single dose of Vc1.1 (1000 nM) caused significant inhibition of colonic nociceptors from healthy mice (***p<0.001, n=10, paired t-test). (ii) Prior incubation with the selective GABABR antagonist CGP-55845 (5 μM) prevented the Vc1.1-induced inhibition of healthy colonic nociceptors (NS, n=5, one-way ANOVA). (E) (i) CVH colonic nociceptors were also inhibited by a single high dose (1000 nM) of Vc1.1 (***p<0.001, n=7, paired t-test). (ii) Prior incubation of CGP-55845 (5 μM) also prevented the Vc1.1-induced inhibition of CVH nociceptors (NS; n=9, one-way ANOVA). ANOVA, analysis of variance.
We then asked if the archetypal GABABR agonist, baclofen, inhibited colonic nociceptors. Baclofen caused a dose-dependent inhibition of colonic nociceptors from both healthy (figure 4Bi) and CVH (figure 4Bii) mice. Interestingly, and similarly to Vc1.1, baclofen also inhibited CVH colonic nociceptors to a greater degree (figure 4C). To confirm that inhibition of colonic nociceptors by Vc1.1 was mediated by the GABABR, we first administered the selective GABABR antagonist CGP55845. In the presence of CGP55845, Vc1.1 no longer inhibited colonic nociceptors from either healthy (figure 4D) or CVH (figure 4E) mice. Finally, we confirmed the expression of GABABR subunits GABABR1 and GABABR2 in colonic DRG neurons by using immunohistochemistry. More than 80% of colonic DRG neurons expressed both GABABR1 and GABABR2 subunits (figure 5A and see online supplementary figure S3). Taken together, these data suggest that the antinociceptive action of Vc1.1 on colonic afferents is mediated via GABABR expressed on colonic afferents.

Colonic dorsal root ganglion (DRG) neurons express γ-aminobutyric acid receptor B (GABABR) subunits and the voltage-gated calcium channels CaV2.2 and CaV2.3. (A) Immunohistochemistry for (i) GABABR1 and (iii) GABABR2 in frozen sections of thoracolumbar DRG from mice that had previously undergone colonic retrograde tracing with CTB-555. A high percentage of traced colonic DRG neurons from both healthy and chronic visceral hypersensitivity (CVH) mice express (ii) GABABR1 and (iv) GABABR2, respectively (healthy: n=6; CVH: n=5). (B) In separate experiments healthy and CVH mice underwent retrograde tracing from the colon with CTB-555. After 4 days thoracolumbar DRG neurons were dissociated and individual colonic DRG neurons were isolated for single-cell-PCR analysis. Gel electrophoresis indicates individual colonic DRG neurons from healthy and CVH mice and their respective expression of GABABR1, GABABR2, CaV2.2 and CaV2.3. Bath controls, perfusate collected during the isolation of single cells, show no expression of any of the targets or reference genes. (C) A high proportion of colonic DRG neurons from (i) healthy and (ii) CVH mice express mRNA for GABABR1, GABABR2, CaV2.2 and CaV2.3. (D) (i) Coexpression of CaV2.2 and GABABR mRNA is found in the majority (>85%) of thoracolumbar colonic DRG neurons from healthy and CVH mice. (ii) mRNA for the GABABR, CaV2.2 and CaV2.3 are coexpressed in the majority (80%) of colonic thoracolumbar DRG neurons from healthy and CVH mice. (E) Quantitative-reverse-transcription-PCR from isolated and pooled (200) colonic DRG neurons shows (i) a significant upregulation of the CaV2.2 exon-37a splice variant in both thoracolumbar and lumbosacral colonic DRG neurons from CVH mice (*p<0.05; healthy: n=4; CVH: n=4). (ii) There is no overall change in total CaV2.2 (exon-37a+37b) levels.
Coexpression of GABABR and CaV2.2 and CaV2.3 in mouse colonic DRG neurons
Recent studies in mammalian cell lines show that Vc1.1-induced inhibition via the GABABR also requires the presence of either CaV2.235 ,39 or CaV2.3.40 To examine whether these channels contribute to the Vc1.1-induced inhibition of colonic afferents, we determined their expression profile in both the TL (splanchnic) and LS (pelvic) pathways innervating the colon. Using qRT-PCR, we found that GABABR1, GABABR2, CaV2.2 and CaV2.3 were abundantly expressed in both mouse TL and LS DRG (see online supplementary figure S4). As colonic DRG neurons represent only approximately 5% of the neurons in these ganglia, we performed retrograde tracing to identify colonic innervating neurons.10 ,12 ,34 Single-cell-RT-PCR analysis and immunohistochemistry determines the expression and importantly the coexpression of these targets specifically within colonic DRG neurons (figure 5B–D, see online supplementary figures S5 and S6). Immunohistochemistry demonstrated that the vast majority of colonic DRG neurons express CaV2.2 (see online supplementary figure S5) and CaV2.3 (see online supplementary figure S6). Single-cell-RT-PCR confirmed the majority of colonic DRG neurons expressed GABABR, CaV2.2 and CaV2.3 (figure 5Ci,ii), with 85% of colonic DRG neurons from healthy and CVH mice coexpressing high levels of GABABR and CaV2.2 (figure 5Di), with 80% coexpressing all three targets, GABABR, CaV2.2 and CaV2.3 (figure 5Dii).
Recent studies indicate there are two isoforms of CaV2.2: exon-37a and exon-37b.41 ,42 Using isoform-specific primers and qRT-PCR from pooled colonic DRG neurons, we found a significant increase in the CaV2.2–exon-37a variant in CVH mice in both TL and LS pathways (figure 5E). This upregulation may explain the increased efficacy of Vc1.1 in CVH states.
CaV2.2 and CaV2.3 contribute to Vc1.1-mediated inhibition of mouse colonic nociceptors
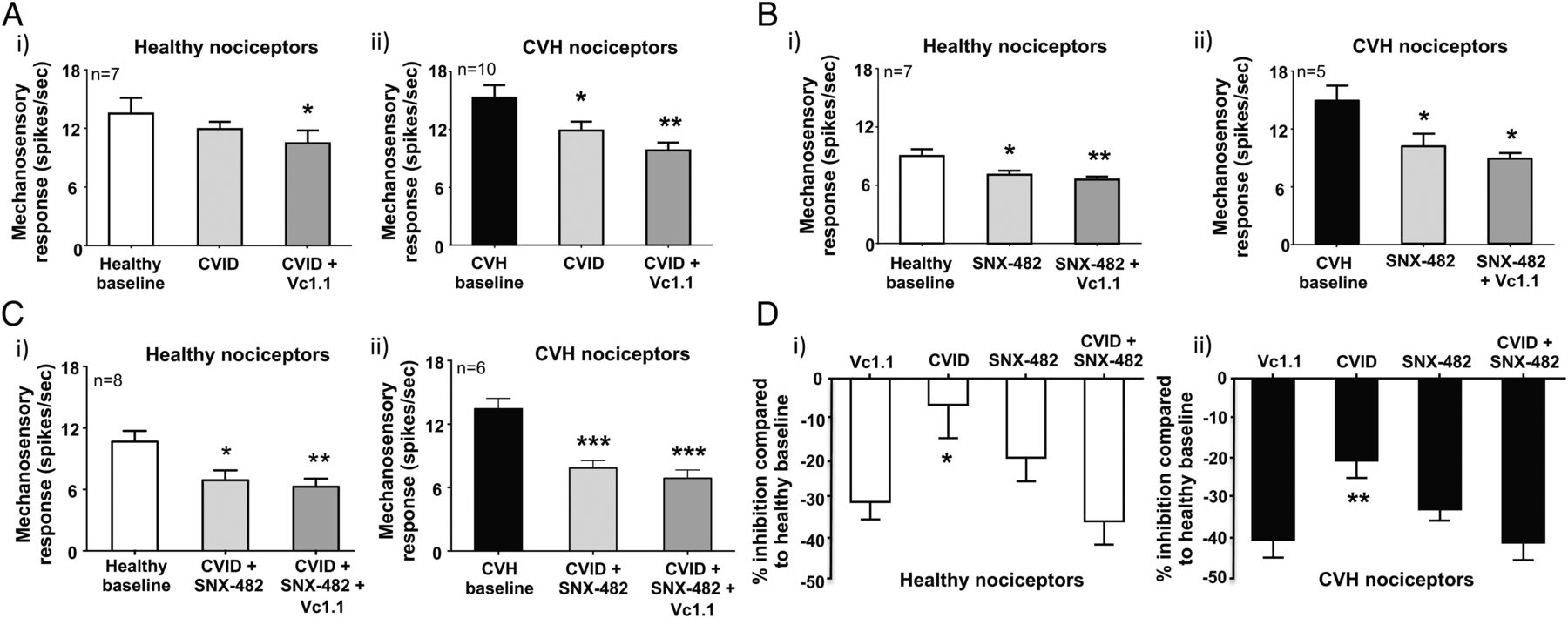
Recombinant cell line studies indicate Vc1.1-mediated activation of GABABR results in the downstream inhibition of both CaV2.2 and CaV2.3 via second messenger pathways.35 ,40 To determine how Vc1.1 inhibits colonic nociceptors, we hypothesised blocking CaV2.2 and CaV2.3, either alone or in combination with maximally effective concentrations of toxin blockers, should also inhibit mouse colonic nociceptors. Using either a selective CaV2.2 (ω-conotoxin CVID) or CaV2.3 (SNX-482) blocker inhibited healthy nociceptors (figure 6Ai,Bi, see online supplementary figures S7A and S8A) and caused greater inhibition of CVH nociceptors (figure 6Aii,Bii, see online supplementary figures S7B and S8B). In separate experiments a combination of CVID and SNX-482 caused pronounced inhibition of healthy nociceptors (figure 6Ci, see online supplementary figure S9A) and even greater inhibition of CVH nociceptors (figure 6Cii, see online supplementary figure S9B). Application of Vc1.1 in the presence of both CVID and SNX-482 had little additional inhibitory effects in both states (figure 6Ci,ii, see online supplementary figures S9A and S9B). Overall, these findings suggest Vc1.1-induced activation of GABABR results in the downstream blockade of CaV2.2 and CaV2.3, which inhibits colonic nociceptor excitability (figures 6D and 7).
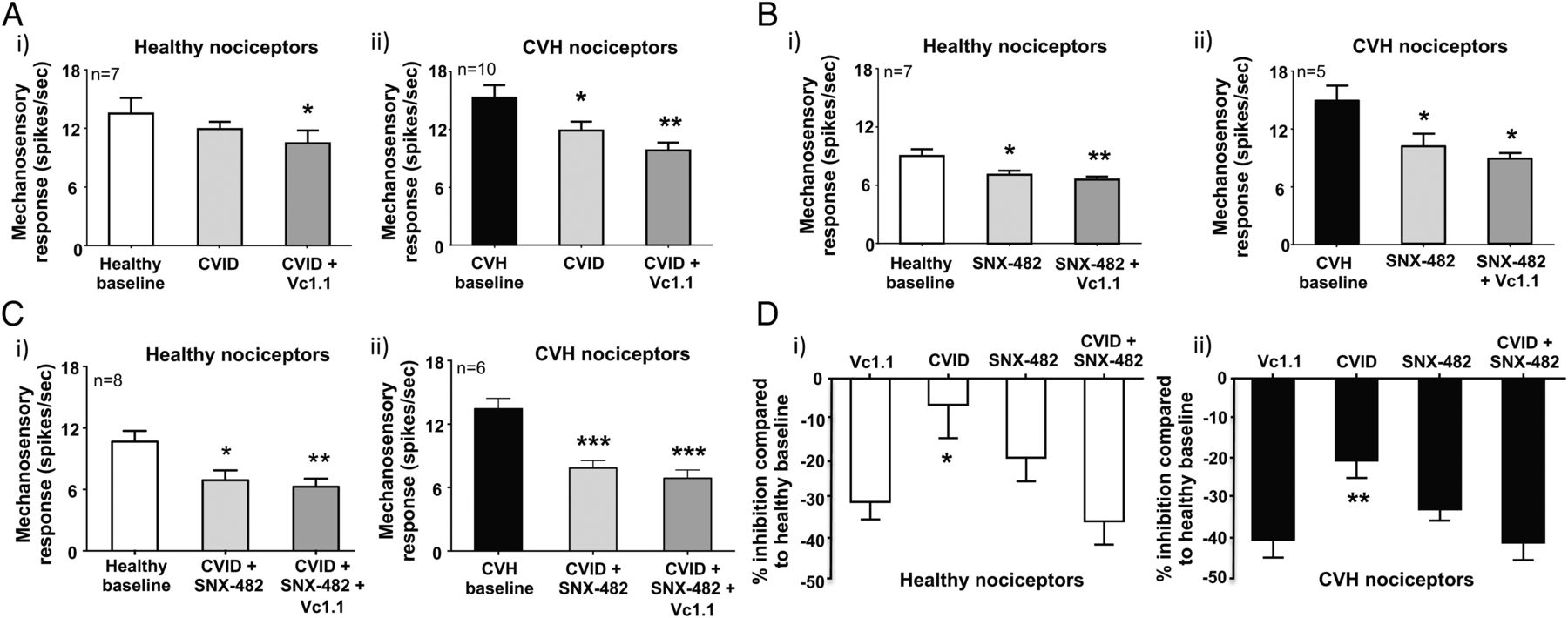
Vc1.1-induced inhibition can be replicated by blocking both CaV2.2 (CVID) and CaV2.3 (SNX-482). (A) (i) In ex vivo preparations colonic nociceptors from healthy mice are inhibited following incubation of the CaV2.2 antagonist CVID (1 μM), although this effect is not significant, whereas (ii) chronic visceral hypersensitivity (CVH) colonic nociceptors are significantly inhibited by CVID (*p<0.05, n=10). In both healthy and CVH colonic nociceptors, the subsequent application of Vc1.1 (1000 nM) in the presence of CVID caused further inhibition (healthy: *p<0.05, n=10; CVH:**p<0.01, n=10). (B) The CaV2.3 blocker SNX-482 (200 nM) inhibited both (i) healthy (*p<0.05, n=7) and (ii) CVH (*p<0.05, n=5) splanchnic colonic nociceptor mechanosensitivity. In both healthy and CVH states the subsequent application of Vc1.1 (1000 nM) in the presence of SNX-482 (200 nM) caused further inhibition of healthy colonic nociceptors (**p<0.01, n=7). (C) The combined application of the CaV2.2 and CaV2.3 blockers, CVID (1 μM) and SNX-482 (200 nM), respectively, significantly inhibits both (i) healthy (*p<0.05, n=8) and (ii) CVH (***p<0.001, n=6) colonic nociceptors with subsequent application of Vc1.1 (1000 nM) causing little additional inhibition. (D) Inhibition of (i) healthy and (ii) CVH colonic nociceptors by single doses of Vc1.1 (1000 nM), CVID (1 μM) and SNX-482 (200 nM) or in the presence of a combination of CVID (1 μM) and SNX-482 (200 nM) expressed as percentage inhibition from healthy nociceptor baseline. CVID causes significantly lesser inhibition than Vc1.1 in (i) healthy (*p<0.05) and (ii) CVH (**p<0.01) nociceptors. However, blocking both CaV2.2 and CaV2.3 in combination with CVID (1 μM) and SNX-482 (200 nM) causes similar inhibition to Vc1.1 alone.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed antinociceptive mechanism by which α-conotoxin Vc1.1 activates γ-aminobutyric acid receptor B (GABABR). Vc1.1 activates GABABR expressed on human dorsal root ganglion (DRG) neurons and on colonic afferent endings resulting in Gβγ and c-Src kinase-mediated inhibition of CaV2.2 and CaV2.3 and subsequent inhibition of neuronal excitability. Modified from Adams et al.14
Discussion
This study provides evidence that the α-conotoxin Vc1.1 inhibits human DRG neurons via activation of the GABABR. It also demonstrates that the peripheral administration of Vc1.1 in mice strongly inhibits the processing of nociceptive information within colonic sensory pathways. We show that both human DRG neurons and mouse colonic DRG neurons express the molecular targets of Vc1.1, the GABABR and its downstream effector channels CaV2.2 and CaV2.3. Correspondingly, we show that Vc1.1 inhibits colonic afferents in both the splanchnic and pelvic pathways and that blocking CaV2.2 and CaV2.3 causes inhibition comparable with that of Vc1.1 alone. These findings highlight the potential therapeutic value of Vc1.1 in the treatment of CVP.
Vc1.1 inhibits sensory DRG neurons, which are the primary transducers of nociceptive information at the start of the pain pathway
A crucial finding of this study was Vc1.1's ability to inhibit a subpopulation of human DRG neurons. This indicates Vc1.1 has an antinociceptive effect in these neurons, which is a key discovery for clinical translatability. These findings were complemented with animal studies where we observed significant Vc1.1-induced inhibition of both colonic nociceptors and low-threshold stretch sensitive afferents that can encode into the noxious range. Crucially, we found that these antinociceptive actions were augmented in a mouse model of CVH. These ex vivo findings translate in vivo as, in response to noxious CRD, mice administered intracolonic Vc1.1 have reduced numbers of activated DH neurons within the TL and LS spinal cord. This finding indicates, in the presence of Vc1.1, a reduced capacity to detect and signal nociceptive events from the colon into the central nervous system. In particular, we observed fewer activated neurons within the superficial lamina of the DH. This is the major termination zone for nociceptive afferents and consists of nociception-specific neurons. Importantly, our findings suggest that Vc1.1 reverses the chronic visceral mechanical hypersensitivity evident in our ex vivo and in vivo studies, rather than completely blocking nociceptive responses. This is important as ideal analgesic agents reverse pathological pain, rather than removing protective pain signalling completely.
Vc1.1 activates GABABR on human DRG neurons and on mouse colonic afferents to inhibit nociceptive signalling
In this study, we have demonstrated for the first time that Vc1.1 inhibits human DRG neuronal excitability via GABABR activation. This was evident as [P6O]Vc1.1, which is inactive at GABABR, does not alter neuronal excitability. Similarly, [P6O]Vc1.1 did not affect mouse colonic afferents, whereas the selective GABABR antagonist CGP55845 prevented Vc1.1-induced inhibition of colonic nociceptors, both in healthy and CVH states. To confirm our proposal that Vc1.1 acts via GABABR, we used baclofen, the archetypal GABABR agonist, and showed that it also inhibits colonic nociceptors. Notably, this inhibitory effect was greater during CVH. The significance of this finding is fourfold. First, these findings closely match those with Vc1.1 and conclusively demonstrate that activation of GABABR on the peripheral endings of colonic DRG neurons within the colon wall results in nociceptor inhibition. Second, although it is known that baclofen inhibits vagal afferents in the upper gut,43 and low-threshold distension sensitive pelvic colonic afferents,44 it has not been previously shown to inhibit colonic nociceptors, or afferents in a model of CVH. Third, in rats, both oral and intravenous administration of baclofen reduces visceral pain-related pseudo-affective responses to CRD.29 ,30 Fourth, baclofen also reduces colonic inflammation-induced neuronal activation within the spinal cord and the brainstem.45 Overall, these observations are consistent with our current in vitro, ex vivo and in vivo findings on the antinociceptive actions of Vc1.1. In response to noxious colonic stimuli, we show inhibition of both colonic nociceptors and low-threshold afferents and a reduction in neuronal activation to noxious CRD in the TL and LS DH of healthy and CVH mice.
Although GABABR have been localised within the rat and human GI tract,46 ,47 crucially we demonstrate for the first time, definitive expression of both GABABR subunits in human DRG neurons and in colonic DRG neurons from healthy and CVH mice. Taken together these studies demonstrate that activation of GABABR on human DRG neurons reduces nociceptive signalling, while activation of GABABR on the peripheral endings of colonic afferents reduces nociception and visceral pain in both healthy and hyperalgesic states. These are important findings and complement studies in other fields of neuroscience, whereby in pyramidal neurons in the cortex, somatic and dendritic GABAB receptors regulate neuronal excitability via different mechanisms.48 Specifically, these studies show that activation of somatic GABAB receptors leads to a reduction in neuronal output, primarily by increasing the rheobase, whereas activation of dendritic GABAB receptors blocks burst firing, decreasing action potential output.48 Our studies recording from the soma of DRG neurons and primary afferent endings in the colon support these mechanisms, where we have observed Vc1.1 increasing the action potential rheobase and decreasing action potential output, respectively.
Vc1.1 as a novel antinociceptive peptide for the treatment of CVP
Although we have shown that the overall antinociceptive effect induced by baclofen and Vc1.1 are similar, it is clear from other studies that Vc1.1 and baclofen act via different mechanisms, in terms of their binding to GABABR and also their downstream targets. For example, Vc1.1 does not compete with baclofen for binding at the ‘Venus fly trap’ on the GABABR, but is proposed to target the interface between the GABABR subunit ectodomains (figure 7).49 Furthermore, whereas baclofen is able to inhibit several different neuronal calcium channels, including CaV2.1, CaV2.2 and CaV2.3, and activate G-protein-coupled inwardly rectifying potassium channels (GIRK) channels, Vc1.1 is more specific by only acting via CaV2.2 or CaV2.3.26 ,35 ,39 ,40 Because baclofen crosses the blood–brain barrier, some of its previously reported antinociceptive effects may be mediated centrally.50 This presents a problem in terms of its off-target effects, which include centrally mediated neurological side-effects, including dizziness.51 In contrast, we show that peripheral administration of Vc1.1 ex vivo and in vivo reduces nociceptive signalling, suggesting a peripheral mechanism of action. Furthermore, as Vc1.1 is a peptide, if delivered peripherally it is unlikely to cross the blood–brain barrier and therefore may be less likely to cause central side-effects. Notably, Vc1.1 has been tested in human clinical trials for treatment of neuropathic pain.19–21 However, its development was discontinued due to lack of potency at its (at the time) proposed molecular target, the human α9α10–nAChR.52 The emergence of an action mediated via the human GABABR suggests its development for chronic pain treatment could resume. Given our current finding of an enhanced Vc1.1 antinociceptive action during CVH, we suggest it is a novel candidate for the treatment for CVP, particularly as cyclised versions of Vc1.1 have impressive stability and are resistant to proteolysis.17
Human DRG neurons and mouse colonic DRG neurons express CaV2.2 and CaV2.3, the key downstream targets of GABABR activation by Vc1.1
The GABABR is a G-protein-coupled receptor and therefore must couple to downstream channels to exert its inhibitory actions. Previous studies demonstrate Vc1.1-mediated activation of GABABR is coupled to both CaV2.2 and CaV2.3 via Gβγ and c-Src kinase second messenger systems.35 ,40 Cav2.2 channels mediate the neuronal N-type calcium current,49 whereas CaV2.3 channels typically conduct a small proportion of the whole-cell calcium current, known as the R-type calcium current.53 Inhibition of CaV2.2, by either gene knockout or selective channel antagonists, causes analgesia in neuropathic pain models.49 Notably, CaV channels have been demonstrated in several studies to contribute to the rheobase of neurons.54 ,55 While CaV channels have been identified as potential therapeutic targets for treating neuropathic pain,56 little is known about the roles of CaV2.2 and CaV2.3 in visceral pain. Here we show that the vast majority of colonic DRG neurons express mRNA and protein for GABABR, CaV2.2 and CaV2.3. Notably, in colonic DRG neurons from CVH mice, we identified a significant upregulation of the CaV2.2 variant exon-37a. Although the CaV2.2 exon-37a variant is expressed at relatively low levels, this is an important finding as exon-37a has been reported to be highly expressed in nociceptors, where it acts as part of a molecular switch controlling N-type current density and G-protein-mediated voltage-independent inhibition.41 ,42 ,57 Accordingly, upregulation of this variant may contribute to the processes responsible for mechanical hypersensitivity and neuronal hyperexcitability in CVH. Furthermore, this upregulation may help to explain the increased inhibitory effects of both Vc1.1 and the CaV2.2 antagonist, ω-conotoxin CVID, in nociceptors from CVH mice. The high expression levels for CaV2.3 in colonic DRG neurons and a considerable inhibitory action of SNX-482 on colonic nociceptors suggests a key role for CaV2.3 in colonic pain pathways. Although CaV2.3 channels are present in some somatic nociceptors58 and can contribute to somatic pain behaviour via spinal and supraspinal mechanisms,59 ,60 our findings are consistent with previous studies suggesting lower CaV2.3 expression in DRG neurons that innervate the epidermis than those innervating deep tissues.61 As we could not specifically identify colonic innervating DRG neurons from human donors, this may explain why we observed less expression of CaV2.3 in whole human DRG and less human DRG neurons expressing CaV2.3.
In conclusion, our findings demonstrate an antinociceptive action for Vc1.1 in human DRG neurons and in colonic sensory afferents. This antinociceptive action is stronger in a model of CVH than in healthy mice and is mediated via the G-protein-coupled receptor, GABABR, which is abundantly expressed in human DRG neurons and mouse colonic DRG neurons. Because altered visceral sensory function is a hallmark of IBS, Vc1.1 represents a potential novel therapy to reduce nociceptive stimuli from the colon and rectum to the central nervous system. Our findings highlight the potential therapeutic value of Vc1.1 and its optimised analogues62 and also identifies the GABABR as a potential target for the treatment of CVP associated with GI disorders.
Acknowledgments
Dr Andrea Ghetti (Anabios) is acknowledged for discussions on the human patch clamp recordings.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
JC, AMH and SG-C contributed equally.
Contributors SMB, DJA and DJC conceived the study. SMB, DJA, AMH, LG, GP, JZ, PEM and JC designed, conducted and analysed experiments. SG-C and JM also conducted and analysed experiments. DJC synthesised Vc1.1 and associated analogues and assisted with critical revision of the manuscript for important intellectual content. SMB wrote the paper and all authors helped with revising the manuscript.
Funding This work was funded by the National Health and Medical Research Council (NHMRC) of Australia Project Grant #1049928 awarded to DJA, SMB and DJC. AMH received funding via the Australian Research Council (ARC) Discovery Early Career Research Award. DJC is an NHMRC Senior Principal Research Fellow. DJA is an ARC Australian Professorial Fellow. SMB is an NHMRC RD Wright Biomedical Research Fellow.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We have included all data relevant to the submission in the manuscript and the supplementary information.