Article Text

Abstract
Objective The pathogenesis of UC relates to gut microbiota dysbiosis. We postulate that alterations in the viral community populating the intestinal mucosa play an important role in UC pathogenesis. This study aims to characterise the mucosal virome and their functions in health and UC.
Design Deep metagenomics sequencing of virus-like particle preparations and bacterial 16S rRNA sequencing were performed on the rectal mucosa of 167 subjects from three different geographical regions in China (UC=91; healthy controls=76). Virome and bacteriome alterations in UC mucosa were assessed and correlated with patient metadata. We applied partition around medoids clustering algorithm and classified mucosa viral communities into two clusters, referred to as mucosal virome metacommunities 1 and 2.
Results In UC, there was an expansion of mucosa viruses, particularly Caudovirales bacteriophages, and a decrease in mucosa Caudovirales diversity, richness and evenness compared with healthy controls. Altered mucosal virome correlated with intestinal inflammation. Interindividual dissimilarity between mucosal viromes was higher in UC than controls. Escherichia phage and Enterobacteria phage were more abundant in the mucosa of UC than controls. Compared with metacommunity 1, metacommunity 2 was predominated by UC subjects and displayed a significant loss of various viral species. Patients with UC showed substantial abrogation of diverse viral functions, whereas multiple viral functions, particularly functions of bacteriophages associated with host bacteria fitness and pathogenicity, were markedly enriched in UC mucosa. Intensive transkingdom correlations between mucosa viruses and bacteria were significantly depleted in UC.
Conclusion We demonstrated for the first time that UC is characterised by substantial alterations of the mucosa virobiota with functional distortion. Enrichment of Caudovirales bacteriophages, increased phage/bacteria virulence functions and loss of viral-bacterial correlations in the UC mucosa highlight that mucosal virome may play an important role in UC pathogenesis.
- gut mucosa
- virome
- bacteria
- virome metacommunity
- ulcerative colitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Alterations in faecal bacteria and faecal virome have been reported in patients with IBD.
Patients with UC showed an expansion of Caudovirales bacteriophages and Caudovirales species richness in the stool.
What are the new findings?
We demonstrated for the first time that UC is characterised by alterations in the mucosa virobiota with functional distortion.
UC mucosa showed a high abundance of DNA viruses, particularly Caudovirales bacteriophages, but a low Caudovirales diversity, richness and evenness. Gut mucosal inflammation was associated with these alterations.
Abundance of Escherichia phage and Enterobacteria phage was significantly higher in the mucosa of UC than controls.
Bacteriophage functions associated with host bacteria fitness and pathogenicity were markedly enriched in UC mucosa.
Intricate interkingdom correlations between mucosa viruses and bacteria were significantly depleted in UC.
How might it impact on clinical practice in the foreseeable future?
Alterations of mucosal virome in UC may contribute to disease pathogenesis.
Therapeutic approaches involving diminishing the abundance of mucosal Caudovirales bacteriophages, including Escherichia phage and Enterobacteria phage, and reconstituting the gut microbial homeostasis between mucosa bacteria and viruses should be explored.
Introduction
UC, one subtype of IBD, is a remitting and relapsing inflammatory disease affecting the entire large intestine, which usually begins in the rectum and spreads proximally.1 The incidence of UC is increasing in the 21st century, especially in newly industrialised countries.2 3 While the aetiology of IBD remains unclear, it is thought to arise from an aggravated immune response towards the gut microbiota in genetically susceptible individuals.4
The gut microbiota is important for human health5–8 and has been implicated in IBD.9 Compelling evidence suggests that gut microbes play a critical role in disease pathogenesis, while geographic, dietary and ethnic factors impact the microbial composition. Most microbiota studies in IBD have investigated the bacterial microbiota and their relationship with the host in disease pathogenesis. With advance in high-throughput sequencing technologies, the unculturable viral component of the gut microbiota is recognised as important habitants of the gut microbial ecosystem, where the number of viral biological entities is believed to far outcompetes that of the bacterial populations.10 11 The healthy human faecal virome consisted of eukaryote-infecting viruses and bacteriophages that infect bacterial cells, with a predominance of the double-strand DNA Caudovirales.12–15 In health, gut bacteriophages show substantial diversity between individuals and are temporally stable.12 14 They latently infect their bacterial hosts, coevolute with gut bacteria which facilitates drifts in the genetic diversity and infectivity of their phages,16 and when under stress they generate progenies that can infect and kill other bacteria.11
To date, we know very little of the role of viruses in IBD pathogenesis. Others have reported alterations in the faecal virome with an increase of bacteriophages, particularly Caudovirales, in mice and humans with IBD.17 18 To address whether gut viruses play an important role in IBD, understanding the composition and function of mucosal virus in health and disease is critical. In this study, we postulate that viruses populating the gut mucosal lining are significantly altered in UC and they play an important role in mucosal inflammation. Here we enriched virus-like particles (VLP) from the rectum of healthy individuals and patients with UC and performed ultradeep virome metagenomics sequencing and 16S rDNA sequencing to determine mucosal virome, bacteriome and viral-bacterial correlations in subjects with UC and healthy individuals. Sequencing analysis of such broader microbiome will elucidate related microbial changes that allow the expression of protective or causative inflammatory organisms. To the best of our knowledge, this study is the first and largest to date to characterise the mucosal virome in health and UC.
Results
Alterations in the diversity of mucosal virome in UC
We compared the mucosal virome of 63 patients with UC and 48 healthy subjects in Hong Kong. On average, 56 632 558±14 330 713 clean paired-end reads were obtained from the enriched rectal VLP preparations. The mucosal virome composition was investigated at the order, genus and species levels in UC and controls. At the order level, Caudovirales were the predominant bacterial viruses (phages) detected in UC mucosa, while at the genus and species level, Ascovirus and Streptococcus phage, respectively, were the dominant viruses detected in UC mucosa (see online supplementary figure 1).
Supplemental material
At the species level, UC subjects showed decreased mucosal virome diversity and richness compared with healthy subjects (t-test, p=0.031 and 0.006, respectively, see online supplementary figure 2). We next explored alterations in the most dominant mucosal viruses, Caudovirales bacteriophages, and found that Caudovirales abundance was significantly higher in UC (Mann-Whitney test, p=0.003, figure 1A), while species diversity, evenness and richness within the Caudovirales order were decreased, when compared with controls (t-test, p=0.009, 0.017 and 0.018, respectively, figure 1B). Collectively, these findings indicate dysbiosis of the mucosal virome in patients with UC.
Supplemental material

Mucosal virome alterations in abundance and diversity in UC. (A) Comparison of Caudovirales abundance in the rectal mucosa of controls and UC subjects. The Caudovirales abundance was calculated as RPKM sum of Caudovirales contigs recruited reads normalised by sequence depth in each subject. The dots indicate individual values of the studied subjects. Statistical significance was determined by Mann-Whitney test, *p<0.01. (B) Comparison of the mucosal Caudovirales α diversity based on Shannon diversity, evenness, Chao1 richness index in the mucosa of controls and UC subjects. Statistical significance was determined by t-test. *P<0.05; **P<0.01. For box plots, the boxes extend from the first to the third quartile (25th to 75th percentiles), with the median depicted by a horizontal line. RPKM, reads per kilobase per million.
Inflamed mucosa showed more enrichment of total viruses and Caudovirales bacteriophages than non-inflamed mucosa of patients with UC (Mann-Whitney test, p=0.015 and 0.017, see online supplementary figure 3A,C). Compared with healthy control mucosa, inflamed mucosa of patients with UC also showed a significant decrease in diversity, evenness and richness of total viruses and Caudovirales bacteriophages (Mann-Whitney test, all p<0.05, see online supplementary figure 3B,D). In contrast, there was no difference in the diversity of mucosal virome between non-inflamed mucosa of patients with UC and healthy control mucosa (see online supplementary figure 3B,D). Overall these findings suggest that gut mucosal inflammation is associated with exacerbated mucosal virome dysbiosis in UC.
Supplemental material
Distinct mucosal virome configurations in UC
Using non-metric multidimensional scaling (NMDS) analysis, we assessed the dissimilarity in the mucosal viral communities between UC and healthy controls. We found that mucosal viromes of UC subjects were significantly different from those of healthy controls at the species level (based on Bray-Curtis dissimilarities, adonis test p=0.001, figure 2A). While disease explained 4.3% of the virome composition variance, discrepancies in virome structure between health and UC were both prominent in the two NMDS axes (t-test, p=0.005 and 0.0005, respectively, for comparisons at MDS1 and MDS2, figure 2B). In addition, viral community dissimilarity among patients with UC was significantly higher than that of within healthy individuals (t-test, p<2.2e-16, figure 2C). These data suggest that virome dysbiosis in UC is highly individualised.

Between-group and within-group viral community structure difference in health and UC. (A) Mucosal viral community structure difference between controls and UC by NMDS (non-metric multidimensional scaling) plotting based on Bray-Curtis dissimilarities at the viral species level. (B) Between-group viral community structure difference reflected on each axis of NMDS plotting. (C) Comparison of within-group mucosal virome Bray-Curtis dissimilarities between healthy individuals and UC subjects. Statistical significance was determined by t-test. **P<0.01; ***P<0.001; ****P<0.0001. For box plots, the boxes extend from the first to the third quartile (25th to 75th percentiles), with the median depicted by a horizontal line.
We next performed DESeq analysis to identify differentially present taxa between UC and controls, at family, genus and species levels. More taxa were found to be enriched in UC than in control mucosa (figure 3). At the family level, Microviridae (single-stranded DNA phage), Myoviridae, Podoviridae (double-stranded DNA phages from the Caudovirales order) and Pneumoviridae (eukaryotic virus) were more abundant in UC than in controls, whereas Anelloviridae (eukaryotic virus) were higher in controls than in UC (figure 3A, see online supplementary figure 4). At the genus level, Phix174microvirus, P1virus, Lambdavirus, T4virus, P22virus (all Caudovirales bacteriophages) and Orthopneumovirus were enriched in UC, whereas giant viruses Coccolithovirus, Minivirus and vertebrate-infecting virus Orthopoxvirus (all eukaryotic viruses) were enriched in controls (figure 3B, see online supplementary figure 4). Consistent with these findings, there was higher abundance of Caudovirales bacteriophage species, including Escherichia phage and Enterobacteria phage in UC relative to control mucosa (figure 3C). Altogether, these findings indicate that dysbiosis in prokaryotic viruses, particularly Caudovirales bacteriophages, is prominent in UC mucosa, while some eukaryotic viruses are more abundant in healthy subjects, suggesting an association between bacteria dysbiosis and bacteriophage expansion in UC.
Supplemental material

Differential viral taxa between health and UC mucosa at the family, genus and species levels. Differentially enriched viral families (A), genera (B) and species (C) between health and UC mucosa were determined by DESeq analysis with false discovery rate (FDR) correction (only those differential taxa with adjusted p<0.05 and |Log2 (between-group fold change)| >2 are shown). For viral taxon names, taxa colour coated by black denote prokaryotic viruses, while those colour coated by orange denote eukaryotic viruses. For viral abundance box plots, the boxes extend from the first to the third quartile (25th to 75th percentiles), with the median depicted by a horizontal line. RPKM, reads per kilobase per million mapped reads.
Recent studies have demonstrated geographical effects on the gut microbiome composition.19 20 We included 56 additional ethnically Chinese subjects from two different regions in China (Beijing, n=40; Xiangshan, Zhejiang province, n=16; see online supplementary figure 5A), to investigate differences between mucosal viromes of healthy controls and UC subjects. Among the disparate mucosal virus species seen between UC and healthy subjects from the Hong Kong cohort, we were able to replicate two virus species in the additional cohort, Chrysochromulina ericina virus and Mimivirus (see online supplementary figure 5B,C), suggesting a geographical effect on the mucosal virome variation. The overpresence of algae-infecting virus C. ericina virus in UC may indicate a detrimental effect of alga-related diet on IBD. Meanwhile, the high presence of the giant virus Mimivirus in UC, a potential pneumonia-causing agent,21 22 is in line with the observation that patients with IBD may be at an increased risk of opportunistic infections including pneumonia.23
Supplemental material
Mucosal virome metacommunities in health and UC
Studies have shown bacterial microbiomes clustering in relation to IBD.24 25 With the observed high degree of dissimilarity among mucosal viral communities, we investigated clustering of mucosal viromes in health and UC. By applying partition around medoids clustering algorithm on the species abundance profile, we found that the viral communities converged into two clusters (referred to as mucosal virome metacommunities hereafter, figure 4A). Among the 85 metacommunity 1 clustered subjects, 40 (47%) were patients with UC. In contrast, among the 26 metacommunity 2 clustered subjects, 23 (88%) were patients with UC (figure 4A), indicating a significant association between patients with UC and metacommunity 2 virome configuration (χ2 test, p=0.0002). We then implemented DESeq and Random Forest analyses to delineate the viral species discriminating these two metacommunities. Forty-six species were discerned between metacommunity 1 and metacommunity 2 viromes, among which 20 species were highly present, whereas 26 were significantly depleted in metacommunity 2 (figure 4B). In metacommunity 2, Escherichia phage, Enterobacteria phage and uncultured Caudovirales phage were the most abundant distinctive species with large effect sizes.
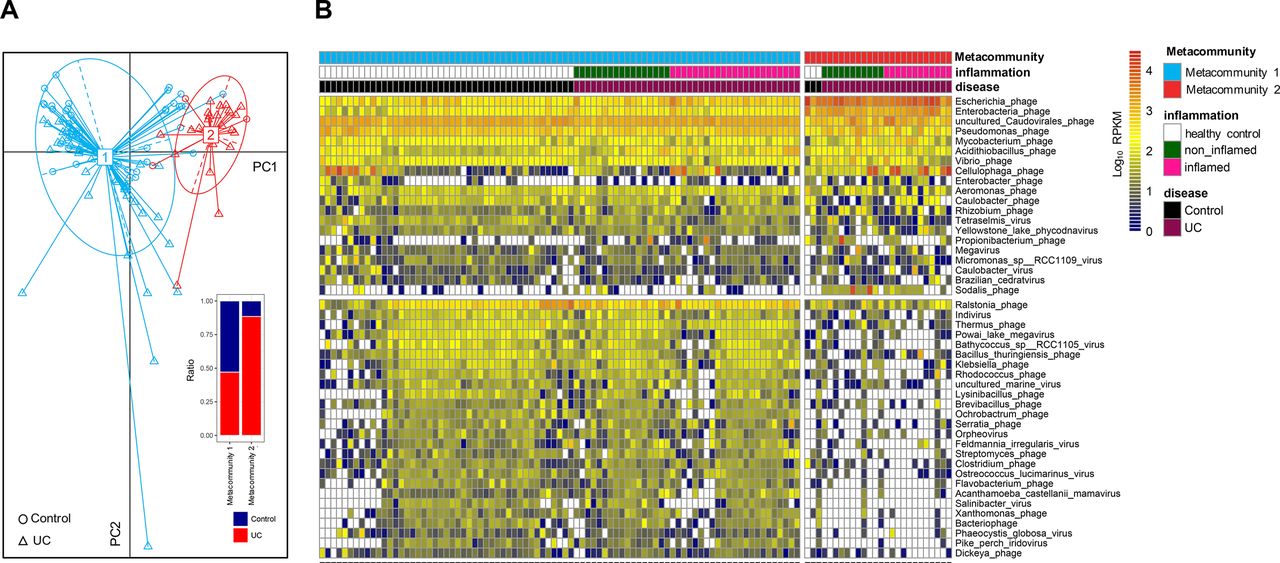
Mucosal virome metacommunities in health and UC. (A) Virome metacommunity clustering based on partition around medoids (PAM) algorithm and principal coordinates analysis (PCoA) on the viral community structures of health and UC mucosa. The inset shows the ratio of healthy individuals and UC subjects within each metacommunity population. (B) Heatmap of the presence of differential viral species contributing to clustering of the two mucosal virome metacommunities. Discriminative species were identified by concordant DESeq and Random Forest analyses. Viral species abundances are colour intensified according to Log10 RPKM values. Only those species concordantly determined by DESeq and Random Forest algorithm with effect size more than 2 and FDR-adjusted p<0.05 are shown. RPKM, reads per kilobase per million mapped reads.
To explore the differences in the mucosal virome composition between healthy individuals and different UC metacommunities, we quantified the presence ratio of viral core species, common species and unique species26 (corresponding to viral species shared among >50%, 20%–50% and <20% of the individuals, respectively) in all study subjects. While no significant differences were seen in the ratio of core species, common species or unique species between healthy and UC metacommunity 1 individuals, the core species accounted for a significant higher proportion of the constituent viral species in UC metacommunity 2 individuals than controls and UC metacommunity 1 individuals (all p<0.0001, one-way analysis of variance [ANOVA], figure 5A,B). However, both common and unique species proportion were diminished in UC metacommunity individuals compared with controls (p<0.0001 and p<0.01, respectively, one-way ANOVA, figure 5A,B). We then assessed the abundance of these viral species, and found that while several top core species were more abundant in UC metacommunity 2 individuals, common species and unique species were decreased or abrogated in UC metacommunity 2 individuals, compared with healthy controls and UC metacommunity 1 individuals (figure 5c).

Significant altered presence of core, common and unique viral species in different UC metacommunities. (A) The proportion of core, common and unique viral species in each subject. The core species, common species and unique species correspond to viral species shared among >50%, 20%–50% and <20% of studied subjects, respectively. (B) Quantification of the presence ratio of core, common and unique viral species in the viral communities of healthy controls and UC subjects with two respective mucosal virome metacommunities. Statistical significance was determined by one-way analysis of variance (ANOVA). **P<0.01; ****P<0.0001. (C) Heatmap of the abundances of the most abundant 100 core, common and unique species in healthy controls and UC subjects with two respective mucosal virome metacommunities. Viral species abundances are colour intensified according to Log2 reads per kilobase per million (RPKM) values.
Exploiting Random Forest, we performed classification on the mucosal viromes of the subjects from Beijing and Xiangshan cohorts. All subjects were classified as metacommunity 1. Differential analysis on all metacommunity 1 subjects between healthy controls and UC in Hong Kong cohort identified a panel of distinctive virus species (see online supplementary figure 6A). However, only two species were replicated, algae-infecting Feldmannia species virus, significantly higher in UC metacommunity 1 subjects relative to control metacommunity 1 subjects in Xiangshan cohort, and Pseudomonas virus, significantly higher in control metacommunity 1 subjects relative to UC metacommunity 1 subjects in Beijing cohort (see online supplementary figure 6B). Altogether, these data highlight a geographical effect on the mucosal virome structure, which may lead to intercohort variations in mucosal virome composition and likely relates to external factors including diet and living environment.
Supplemental material
Functional alterations of mucosal virome in UC
To assess functions of mucosal virome, we performed HUMANN2 analysis on the viral sequences against Gene ontology (GO) and Pfam protein family databases. Due to the largely underexplored nature of gut mucosal virome functions, we were only able to characterise the function of a small proportion of the viral reads. Among the annotated functions, integral component of membrane, DNA binding (GO terms), ATP-binding cassette (ABC) transporter and integrase core domain (Pfam proteins) were the prominent proteins/functions (see online supplementary figure 7), suggesting an active role for viruses in virus-host biology and significant interplay between gut viruses and bacteria.
Supplemental material
We then compared the predicted mucosal virome functions between health and UC. Though the most abundant functions did not differ in abundance between health and UC, healthy individuals showed a richer virome function, in terms of GO and Pfam protein functions, whereas UC subjects exhibited a significant abolishment of functions (figure 6). Nonetheless, several molecular functions were determined to be more abundant in UC than in healthy mucosal virome, including DNA template negative regulation of transcription, beta-lactamase, glutamine amidotransferase, glycosal hydrolases, type II/IV secretion system and multicopper oxidase, all of which were linked to phage lysis of bacteria host as well as functions of bacteria origin. This result implies that the enriched mucosal viral functions in UC were associated with bacteria fitness, pathogenicity and antibiotics resistance.

Significant loss of diverse viral functions in UC mucosa with concomitant increases in bacteria-pathogenicity associated functions. (A) Presence-absence heatmap of the classified viral functions in controls and UC. Viral functions were predicted and classified via HUMANN2 pipeline, exploiting the sophisticated Gene ontology (GO) and Pfam protein family databases. Functions with reads per kilobase (RPK) >10 were considered present in individuals. The abundance distribution of classified viral functions is plotted in the line chart, with abundance values expressed as Log2 RPK. (B) Differentially enriched viral functions between health and UC mucosa. Differential viral functions were determined by DESeq analysis with FDR correction. Only those functions with adjusted p<0.05 and |Log2 (between-group fold change)| >2 are shown. For box plots, the boxes extend from the first to the third quartile (25th to 75th percentiles), with the median depicted by a horizontal line.
Mucosal bacteriome alterations and transkingdom network between virome and bacteriome at the mucosa
We further assessed the bacterial microbiome alteration in UC mucosa compared with healthy control mucosa. Bacterial diversity and richness were significantly increased (t-test, p=0.024 and 0.007, respectively, see online supplementary figure 8A), indicating an expansion of diverse bacteria in the mucosa of patients with UC. UC mucosal bacteriome showed distinctive structure to that of controls, at the phylum, family and genus levels (see online supplementary figure 8B–D). Quantitative differential analysis identified a number of bacterial taxa distinguished between health and UC mucosa (see online supplementary figure 8E). Among them, Firmicutes (phylum), Pseudomonadaceae, Ruminococcaceae, Thermaceae, unclassified Clostridiales, Veillonellaceae (family) and Coprococcus (genus) were significantly enriched in UC than in controls.
Supplemental material
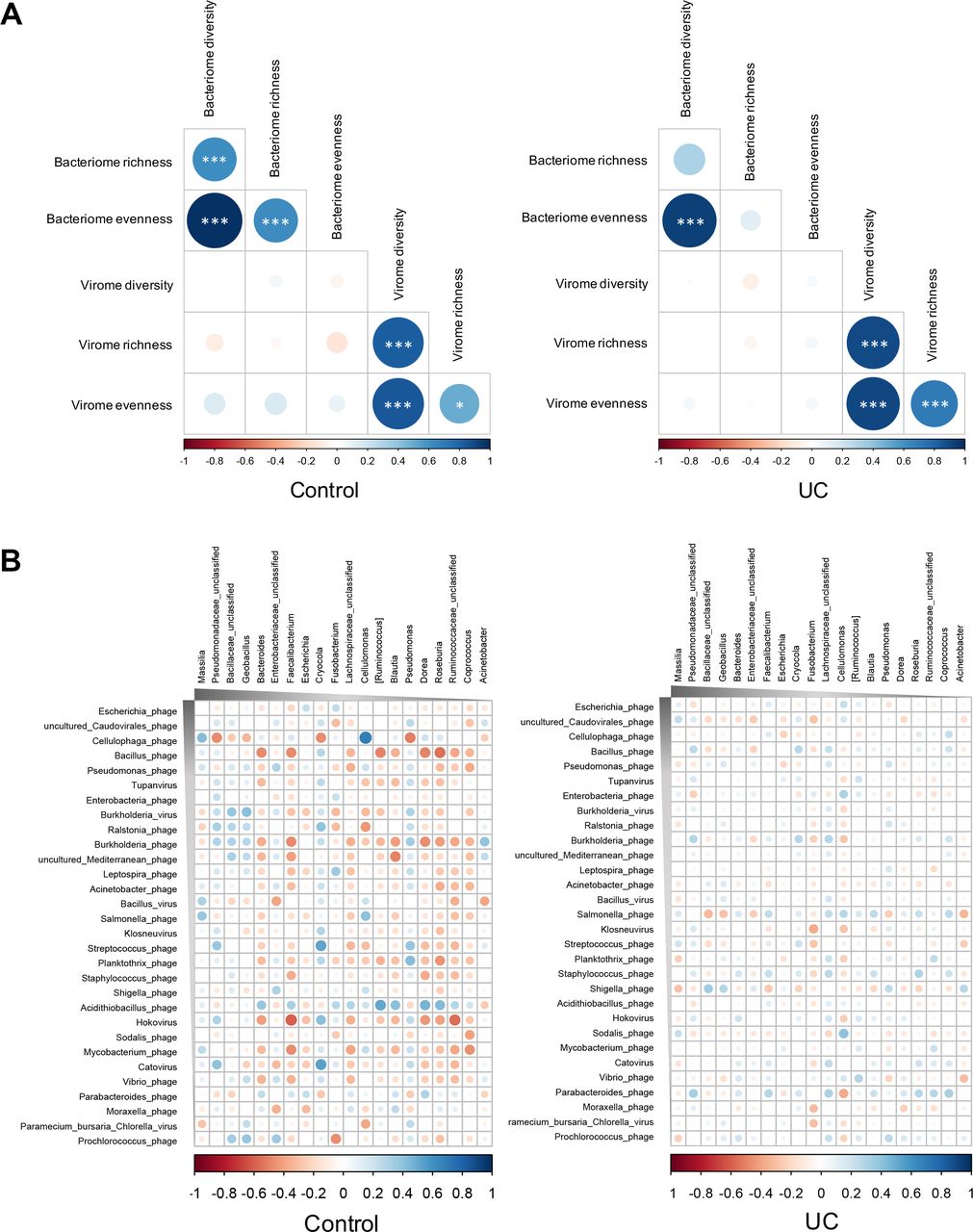
To characterise the relationship between bacteriome and virome in the mucosa, we evaluated the correlation between the α diversity (diversity, evenness and richness) of the bacteriome and virome. In controls, there were significant correlations between intrakingdom α diversity metrics (figure 7A). However, correlations of within-bacteria-kingdom α diversity seen in controls were lost in UC (figure 7A), suggesting a more dysbiotic state of the bacterial microbiota than the virobiota in UC mucosa. We then assessed the correlation between bacterial genera and viral species in control and UC mucosa. The alterations of viral-bacterial correlations in UC could be ascribed to both losses of viral-bacterial associations and appearances of novel viral-bacterial associations. A larger number of bacterial-viral correlations were seen in UC than in controls (figure 7B). Among these bacterial-viral correlations, most were negative correlations. However, a substantial contraction in the number of significant bacterial-viral correlation was observed in UC mucosal samples, meanwhile the correlations in UC were weakened compared with that in controls (figure 7B). In particular, the correlations between the virus species Cellulophaga phage, Bacillus phage, Burkholderia phage, Acidithiobacillus phage (all Caudovirales bacteriophages), Hokovirus and mucosal bacteria were extensively intensive in controls, whereas they were significantly dampened in UC. This corresponded with an expansion in the abundance of Cellulophaga phage and Acidithiobacillus phage in patients with UC, along with a depletion of correlations between these two species and mucosal bacteria. These results suggest an altered virobiota–bacterial microbiota relationship in UC, where viruses and bacteria became less intertwined and more specialised, further implicating the importance of interkingdom equilibrium among mucosal viruses and bacteria for human health.
![[gutjnl-2018-318131supp001.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
![[gutjnl-2018-318131supp002.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-318131supp003.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-318131supp004.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-318131supp005.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
{kind=link}
![[gutjnl-2018-318131supp006.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
![[gutjnl-2018-318131supp007.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-318131supp008.jpg]](https://gut.bmj.com/content/gutjnl/68/7/1169/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
{kind=link}
Mucosal bacterial-viral correlation patterns in health and UC. (A) Correlations between the α diversity (diversity, evenness and richness) of mucosal bacteria and viruses in healthy controls and UC, respectively. Spearman’s correlation coefficient was calculated, while statistical significance was determined for all pairwise comparisons. Significant correlations (p<0.05) are displayed with asterisk. *P<0.05; ***P<0.001. (B) Correlations between the most abundant 30 virus species and the most abundant 20 bacteria genera in health and UC mucosa. Spearman’s correlation coefficient was calculated, while statistical significance was determined for all pairwise comparisons. Only statistically significant correlations were plotted, where blue circles indicate positive correlations and red circles indicate inverse correlations. The size and shading indicate the magnitude of the correlation where darker shades denote more intensive correlations than light ones.
Discussion
To our knowledge, this study represents the first and most in-depth human mucosal virobiota study in health and UC using a dedicated metagenomics approach based on enriched virome preparation. We showed that patients with UC exhibited a dysbiotic mucosal virobiota, characterised by an increase in virus abundance, particularly Caudovirales bacteriophages, and a decreased viral diversity, richness and evenness. Given the underexplored nature of virome for which the viruses database remains small and incomplete, unknown viruses can influence the estimation of diversity, limiting our effort to disentangle mucosal viromes in health and UC. We also observed significant functional alterations in mucosal virome in UC, which were associated with bacteriophage lytic cycle, and bacteria host features, including bacteria adaption, antibiotics resistance and pathogenicity. Inflammation was associated with shifts in virome composition and alga-infecting viruses were more abundant in UC than controls. We hypothesise that host inflammation and environmental factors such as medication or diet may be significant contributors to the altered mucosal viromes, which are intertwined with the bacterial microbiome and host immune responses.
In mice, it has been shown that augmented bacteriophage populations during colitis were associated with intestinal inflammation in the pathobiont host,18 and inflammation can boost free phage production and subsequent transfer in mice.27 In humans, Caudovirales were shown to be present in high abundance in patients with IBD17 and Clostridium difficile infection.28 In another study, a healthy core phageome was identified in healthy individuals and decreased numbers of these core phages in the faeces were reported in patients with UC.26 More recently, an increased faecal eukaryotic virome richness has been described in patients with UC from a Belgian cohort.29 Increased abundance of Escherichia phage and Enterobacteria phage and decreased core viral species numbers in UC mucosa in this study are consistent with findings from the faeces of colitic mice18 and patients with IBD.17 In this cross-sectional study, we were unable to discern whether alterations in the virome are the cause or consequence of mucosal inflammation and denudation of the epithelial barrier. Future animal studies will be necessary to investigate this further.
A marginal inverse correlation between Escherichia phage and Escherichia genus bacteria in UC mucosa was observed (figure 7B). The enhanced Escherichia phage abundance and antagonistic relationship between Escherichia phage and the bacteria genus Escherichia in UC mucosa, shown in our study, along with reported expansion in both Escherichia phage and Escherichia in UC faeces by others,17 18 may indicate that Escherichia phage contention at the mucosa is linked to coexpansion of both in the faeces, suggesting prophage induction and complex bacteriophage-bacteria dynamics in the gut. It is postulated that apart from the empirical ‘predator-prey’ phage-bacteria dynamics, there are also ‘kill-the-winner’ and ‘arms-race’ phage-bacteria dynamics at play in the gut milieu where coexpansion and coevolution of phages and bacteria are expected, particularly when host is under stress.30
Alterations of bacteriophage composition in the gut mucosa can influence the bacterial microbiota ecology. Bacteriophages are primary drivers of bacterial fitness and diversity.31 Lysis of bacteria could result in a release of nucleic acids, proteins and lipids that serve as pathogen-associated molecular patterns that trigger inflammatory responses to induce cellular infiltration, cytokine production and even tissue damage. In the GI tract, bacteriophages are responsible for horizontal transfer of genetic elements among bacterial populations, including those for antibiotic resistance and disease pathogenesis.32–35
The human virome evolves rapidly due to varying conditions and stressors conferred by the host and the environment. A multitude of factors contributing to the hypervariation in virome relative to the bacteriome among populations have been reported.12 36–39 Disaccordance in discriminative viral species identified between Hong Kong cohort and other China cohorts implies high degree of variation in mucosal virome configuration, likely a region-population-specific effect on the virome structure. However, our study is limited by a modest sample size of the China cohort and future large population-based studies will be important to confirm universal and population-specific virome in health and IBD.
Although diverse bacteriophages were present in high abundance in UC, some eukaryotic viruses were more abundant in healthy individuals. Chronic infections with viruses are common in humans.11 The history of infection with acute and chronic viruses contributes to host immunity education and maturation,40–42 especially when it occurs in early life.2 43 Viral pathogen-driven selection assists in shaping the immune system, improving immune tolerance and memory, thereby preventing against autoimmune disease development.41 44–46
Microbiota composition has been reported to differ between faecal and mucosal samples. Most IBD microbiome studies have focused on faecal samples. There has been controversy over the degree of dysbiosis in IBD, particularly at the mucosal level.47 Studies reporting changes in the diversity, richness and composition of the bacteriome in UC mucosa have produced inconsistent results. Multiple studies have shown decreased bacterial diversity and richness in IBD mucosa,48 49 several other studies however indicated similar levels of bacterial diversity and richness between UC and control mucosa50 51while some on the contrary showed increased bacterial richness in the non-inflamed tissues from IBD subjects compared with healthy subject tissues.52 Though some studies reported overpresentation of Proteobacteria in UC mucosa compared with healthy mucosa,48 53 some reported insignificant difference.50 Additionally, while some studies reported decrease in Firmicutes in UC mucosa compared with healthy mucosa,48 49 some other groups showed higher abundance of Firmicutes in UC mucosa than healthy mucosa,50 53 54 the latter findings are consistent with our data. The heterogeneity observed is likely to be multifactorial including the site of sampling, medication use, disease phenotype, active versus inactive disease, oxidative stress level as well as differences in population, geography and diet, all of which have been documented to influence mucosal bacteria composition. Further large-scale in-depth investigations and validation are necessary.
In conclusion, our findings highlight the importance of mucosal virome in the pathogenesis of UC. A healthy mucosal virome is characterised by a low abundant but diversified viral community, which rests in homeostasis with the host and other microbial communities. Alterations in the mucosal virome in UC may contribute to disease pathogenesis and restoration of the mucosa viral community represents promising therapeutics for UC.
Methods
Cohort description and study subjects
A clinical cohort of 63 patients with UC versus 48 healthy subjects from Hong Kong was included in this study. In addition, 20 healthy subjects versus 20 patients with UC from Beijing, and eight healthy subjects versus eight patients with UC from Xiangshan, Zhejiang Province (all Chinese), were also enrolled into this study, with informed consent. Patient inclusion criteria include subjects aged ≥18 with a diagnosis of UC defined by endoscopy, radiology and histology. Consecutive patients with UC who consented to have colonic samples taken during surveillance colonoscopy or colonoscopy as part of assessment for disease activity were included. Tissue biopsies were obtained from the rectum. Majority of patients were in clinical remission. Controls comprise individuals undergoing colonoscopy for screening for polyp, colorectal cancer, or symptoms of rectal bleeding due to haemorrhoids, and friends and spouses or partners of patients at local hospitals, or any individuals who were interested to participate in this study. Consecutive healthy individuals underwent screening colonoscopy and were found to have a normal macroscopic colon and no evidence of microscopic inflammation after colonoscopy.
Rectal biopsies from the study subjects were collected via endoscopy and then stored at −80°C for downstream mucosal virome and bacterium analysis. Patient metadata were shown in online supplementary table 1.
Supplemental material
VLP enrichment and sequencing
VLPs were enriched from the rectal biopsies of patients with UC and healthy subjects, using a modified protocol according to previously described methods.17 28 32 The detailed procedure is included in the online supplementary appendix.
Supplemental material
VLP DNAs were quantified (Nanodrop) and 1 µg of DNA was randomly fragmented by ultrasonication (Covaris) followed by library construction. The qualified libraries were amplified on cBot to generate the cluster on the flow cell (TruSeq PE Cluster Kit V3-cBot-HS, Illumina). The amplified flow cell was sequenced pair end on the HiSeq Xten System (TruSeq SBS KIT-HS V3, Illumina) (BGI, Shenzhen, China; standard 2×150 bp run), generating 20–60 million raw sequences (5–8G raw data) per sample. The detailed sequence statistics was included in the online supplementary table 2. Sequence processing and quality control, de novo contig assembly and taxonomy annotation were included in the online supplementary appendix.
Supplemental material
Virome data analysis
To estimate contig abundance and calculate sequence diversity, all reads were aligned to the resulting curated contigs using Bowtie2 (V.2.2.9).55 The mapped sequence counts, contig lengths and total sequence counts were used to normalise the sequence counts and represent the reads per kilobase per million of each sample to the contigs. These values were used to generate viral abundance tables at various taxonomy levels.
The virome abundance data were imported into R V.3.2.3. Diversity, evenness and richness calculation were performed using phyloseq in R. Spearman’s correlation and their significance were calculated using the cor and cor.test functions in R, respectively. For the viruses–bacteria correlations in α diversity and taxa abundance, Spearman’s correlations were calculated. Correlation plots were generated using the corrplot R package. Heatmaps were generated using the pheatmap R package. DESeq, Random Forest and LEfSe linear discriminant analysis were included in the online supplementary appendix.
Accession codes
Sequence data have been deposited to the NCBI Sequence Read Archive under BioProject accession numbers PRJNA504921 and PRJNA506811.
References
Footnotes
Contributors TZ performed the experiments and metagenomics analysis and drafted the manuscript. XJL, YZ and WT performed study subject inclusion and collection of specimens, helped in virome preparation and analysis. CPC, SL, FZ and RZ collected the clinical samples and data, helped in virus DNA extraction and provided significant intellectual contribution. WT and JYLC managed the clinical practice and clinical database. PC, JJYS, JY, FKLC, QC and JQS provided intellectual contribution and critical comments on the manuscript. SCN designed and supervised the study and revised the manuscript.
Funding PROCORE-France/Hong Kong Joint Research Scheme (F-CUHK402/15); ANR/RGC Joint Research Scheme (A-CUHK402/17); the ENIGMA study supported by Helmsley Charitable Trust; and a seed fund for Gut Microbiome Research provided by the Gut Microbiota Research Center, Faculty of Medicine (The Chinese University of Hong Kong).
Competing interests None declared.
Ethics approval The study was approved by The Joint Chinese University of Hong Kong, New Territories East Cluster Clinical Research Ethics Committee (The Joint CUHK-NTEC CREC, CREC Ref No: 2014.026).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.