Article Text

Statistics from Altmetric.com
- gastrointestinal neoplasia
- colorectal cancer
- chronic inflammation
- JC virus
- viral oncogenesis
- DNA mismatch repair
- p53
- inflammatory bowel disease
- mutagenesis
- microsatellite instability
INTRODUCTION
The struggle to understand the origins of human cancers has captured the imagination of many investigators. The epidemiology of human cancers and the availability of many laboratory models of cancer could give the casual observer the impression that all cancers are a result of exposures to chemical carcinogens in the environment. This is only part of the story, and in most instances environmental exposures are very different in scale from what is required in laboratory animals to induce tumours. Actually, most laboratory models have been carefully developed to match the carcinogen with the host. For example, several alkylating agents can induce intestinal cancers in rodents.1 However, the distribution of the neoplasms throughout the gut varies from one mouse strain to another, and in some instances the carcinogens induce tumours only outside the gut. Lower doses—perhaps those more relevant for human cancers—may be tolerated, and not induce cancer at all. The mechanisms for tumour development in the human gastrointestinal tract appear to be a much more complicated issue.
This review of recent advances in basic science will focus on newly discovered mechanisms involved in the development of colorectal cancer (CRC). This is potentially a very broad topic, and therefore we have selected two novel mechanisms for emphasis. Firstly, a virus carried by most healthy individuals has been implicated as a possible cause for chromosomal instability (CIN), the process that leads to aneuploidy. Chromosomal aberrations and this form of genomic instability play a major role in the development and progression of multistep carcinogenesis, such as occurs in CRC.2 Secondly, it is abundantly clear that inflammation is carcinogenic, and furthermore, endogenous processes that can modify the ability of the host to cope with inflammation appear to modify the risk of cancer to the host. There is growing evidence that this may be germane to cancer risk in inflammatory bowel disease (IBD). Almost certainly, additional mechanisms will be found in the future but these may be particularly pertinent for CRC, and perhaps cancer elsewhere in the gut.
VIRAL INFECTION AND GASTROINTESTINAL CANCER
There has been gradual acceptance that viruses can participate in the induction of some cancers. This concept is nearly a century old, and dates back to the observation by a New York farmer that his chickens were highly prone to tumours. Subsequent experiments conducted by Peyton Rous eventually revealed that an avian retrovirus carries a mutated proto-oncogene, which is linked mechanistically to these sarcomas.3 Although the links between viruses and cancer are less obvious in humans, it is generally agreed that hepatitis B virus and hepatitis C virus are aetiologically linked to the development of hepatomas. Similarly, human papillomaviruses have been linked to human genital tract tumours by virtue of the coordinated action of two oncogenes encoded by this virus, and vaccination against this virus has begun in the hope of preventing those cancers.
The role of polyomaviruses in human cancer has been much more controversial. Safety studies performed on the human polio vaccine in the early 1960s revealed contamination by SV40 virus, which originated in the simian kidney cells used to culture the poliovirus. Large numbers of humans were exposed to SV40 while receiving their polio vaccines. Although the virus is present in some cancers, evidence for its role in carcinogenesis is inconclusive.4 However, injecting SV40 into rodents clearly causes cancer. The small number of genes encoded by this virus, and the unequivocal oncogenic activity of the T antigen, have raised the possibility that human polyomaviruses may cause cancers in the human host by virtue of encoding a potent oncogene.
There are two viruses closely related to SV40 in which the natural host is humans rather than monkeys. These are JC virus (JCV) and BK virus (BKV). SV40 has evolved into a useful laboratory tool for transforming cells, and its presence in the genome leads to chromosomal aberrations and genomic instability. Based on close structural similarity, it is reasonable to speculate that these human viruses may play a role in human carcinogenesis.
JCV structure and epidemiology
JCV was first isolated from the spinal fluid of a patient suffering from progressive multifocal leucoencephalopathy, or PML.5 JCV belongs to the subfamily of non-enveloped DNA viruses with icosahedral capsids that contain small (40–60 nm), double stranded, negatively supercoiled, circular DNA genomes.6 The 5.13 kb circular genome of JCV encodes only six genes, and has two regions of approximately equal size known as early and late transcription units, as illustrated in fig 1. The early and late coding regions are separated by a bidirectional transcriptional control region (TCR), which is the most divergent DNA sequence among the polyomaviruses JCV, BKV, and SV40. The late region encodes the capsid structural proteins VP1, VP2, and VP3 through alternately spliced mRNAs, and a small regulatory protein known as “agnoprotein” which is involved in viral assembly. The early region encodes the alternatively spliced transforming proteins large T antigen (T-Ag) and small t antigen (t-Ag). T-antigen plays a critical role in the life cycle as it directs viral early and late gene expression, as well as viral DNA replication during lytic infection.7
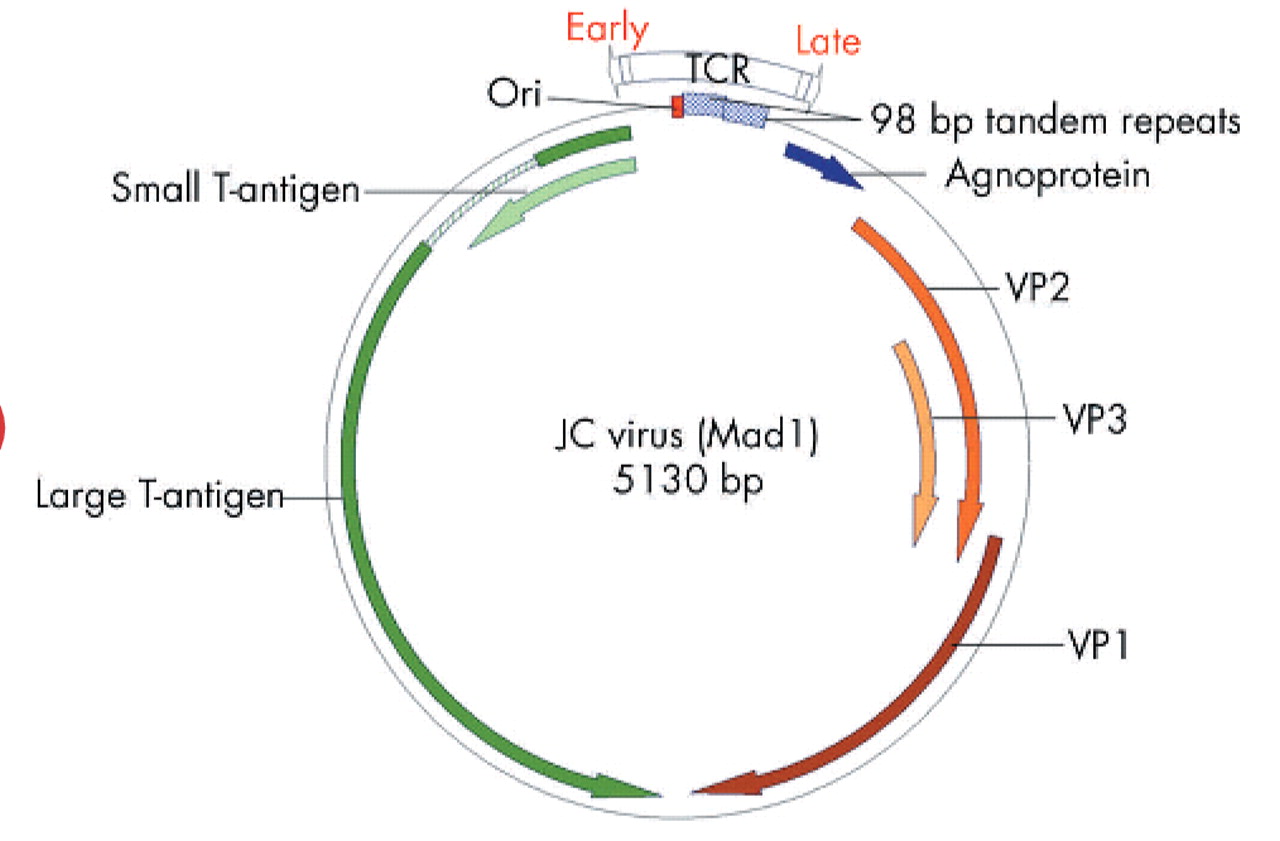
The JC virus (JCV) genome. The JCV genome is small (5.13 kb) and contains a limited coding capacity. There is a non-translated regulatory region known as the transcriptional control region (TCR) of about 400 bp that contains the origin of replication (ori), and the promoters and enhancers that control replication. The early genes (counterclockwise) encode two replication proteins, large T-antigen and small t-antigen, which are expressed soon after the virus enters the cell. The late region (clockwise) encodes the capsid proteins (VP1, VP2, and VP3) and a maturation protein (agnoprotein), and is expressed only after viral DNA replication has begun. The transcriptional promoters and enhancers are located adjacent to the functional ori sequence. The TCR of the Mad-1 strain of JCV contains two 98 base pair tandem repeats which contain binding sites for various transcription factors. Viral transcription is mediated by cellular RNA polymerase II and early and late transcription proceed bidirectionally from near the ori, with the early and late transcripts being produced from opposite strands of viral DNA. Alternative splicing of pre-messenger RNAs produces functional mRNAs. Large T-antigen is not a transcription factor per se but it can autoregulate the early promoter as the replication cycle proceeds. When large T-antigen reaches a sufficient concentration in the cell, it binds to viral DNA which may block the assembly of functional transcriptional complexes, repressing early transcription. Large T-antigen indirectly contributes to activation of JCV late transcription, perhaps by stabilising interactions among transcription factors. Viral DNA replication requires a functional JCV ori, large T-antigen protein with intact DNA binding and helicase activities, and several cellular proteins involved in DNA synthesis. Large T-antigen binds to specific sites in the JCV ori, catalyses local unwinding of the viral DNA, and recruits cellular DNA proteins to the complex, including topoisomerase I and DNA polymerases. Topoisomerase II separates the newly joined replicated circular DNA daughter molecules.
Epidemiological studies have demonstrated that JCV is ubiquitous in the human population, and approximately 60–80% of adults have specific antibodies to JCV. The virus is believed to be transmitted by close contact during early childhood years, and primary infections are typically harmless.8 Reappearance of the virus in the urine of pregnant women has led to the hypothesis that latency may be maintained, at least in part, in the kidney.9 The presence of viral genomic sequences in the gastrointestinal tract and in raw urban sewage suggests possible faecal-oral transmission of JCV, and emphasises the relative stability of the viral particles and the potential for interpersonal spread.10,11 JCV persists indefinitely in both kidney and B lymphocytes. However, under conditions of immunosuppression, as occurs in patients with acquired immunodeficiency syndrome, JCV can emerge from latency to cause PML.12,13 Recently, it was elegantly demonstrated that the serotonergic receptor 5-HT2AR acts as the cellular receptor for JCV in human glial cells, suggesting the possibility that serotonin receptor antagonists may possibly find a role in the treatment of PML or other diseases caused by JCV.14
JCV and human cancers
In addition to its primary role in the development of PML, in recent years mounting evidence has suggested that JCV may be associated with human neoplasms in the absence of immunosuppression or PML. This has been possible through the use of sensitive techniques and the availability of more reliable immunohistochemical reagents, which have allowed the detection of JCV genomic sequences and T-antigen expression in a variety of human malignancies, including brain tumours of glial origin,15 medulloblastomas,16 colon carcinoma,11,17,18 and oesophageal cancers.19,20
Although the aetiological role for JCV in the development of various human malignancies remains to be established, it is believed that the early proteins of the virus, and particularly the T-antigen, play a critical role in malignant transformation by associating with several cellular proteins.21 Similar to SV40 T-antigen, JCV T-Ag is a multifunctional protein that possesses the ability to bind and break DNA, and has helicase and ATPase activities, which are needed for DNA replication.21,22 Furthermore, JCV T-antigen can also dysregulate control of the cell cycle by interacting with the tumour suppressor proteins p53 and pRb, making it unique in its ability to simultaneously disrupt chromosomal integrity and inactivate cell cycle checkpoints. This coordinated action permits the replication and proliferation of cells with damaged DNA (fig 2).19,23 Moreover, T-antigen may distort control of cellular proliferation by deregulating the Wnt signalling pathway through stabilisation of β-catenin,24 interacting with IGF-IR signalling,25 and inducing chromosomal instability in B lymphocytes.26
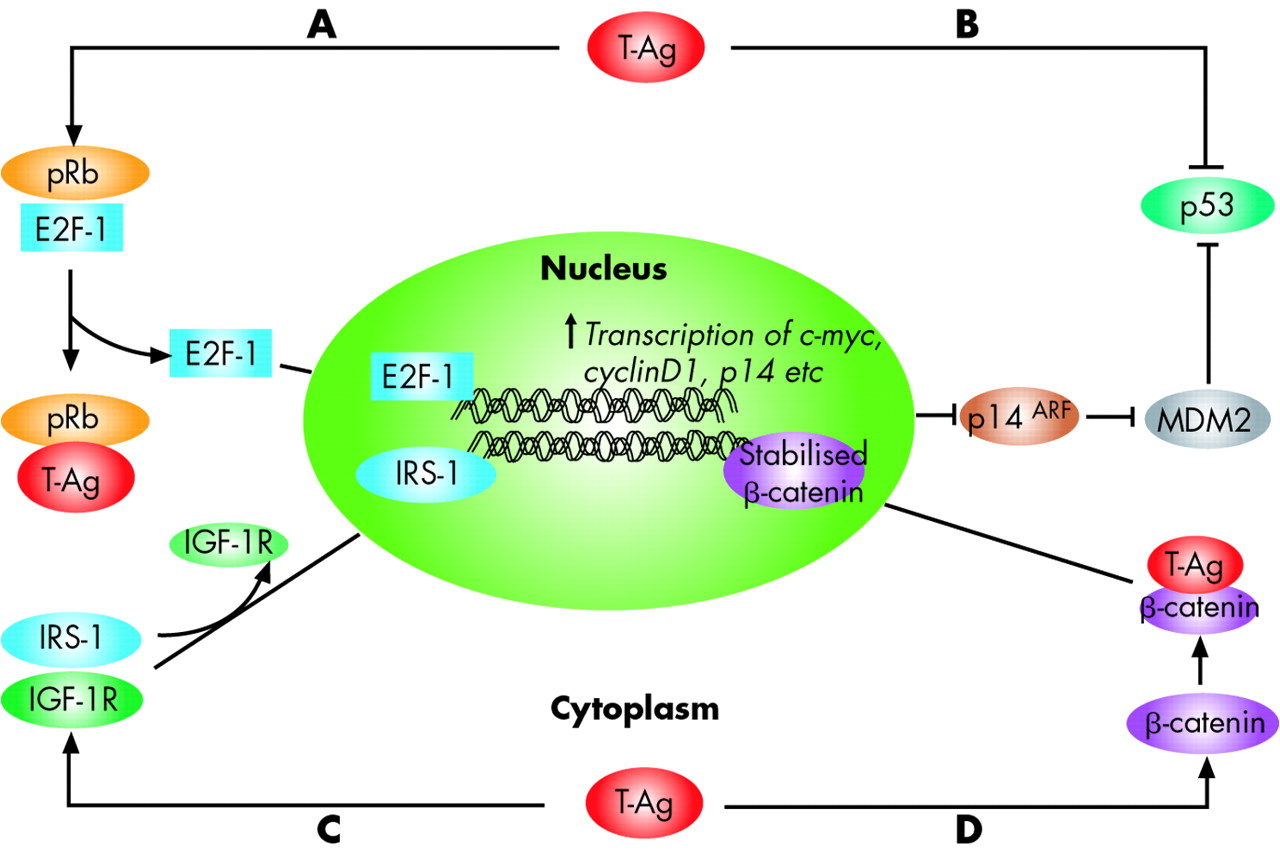
Interaction of JC virus (JCV) T-antigen with key growth regulatory pathways. JCV large T-antigen (T-Ag) is believed to drive the oncogenic process of malignant transformation by associating with several cellular proteins. T-Ag has the capacity to interact with two tumour suppressor proteins that regulate cell cycle progression, pRb and p53. (A) Under normal physiological conditions, pRb is maintained in a hypophosphorylated state and sequesters the S phase specific transcription factor E2F-1, in a pRb:E2F-1 complex. On JCV infection, T-Ag binds to the pRb protein releasing E2F-1, permitting it to induce cellular proliferation by promoting transcription of S phase genes. E2F-1 induces p14ARF gene expression, which interferes with the activity of MDM2, a negative regulator of p53 protein. Consequently, p53 is stabilised, which would inhibit the cell cycle and promote apoptosis (see fig 6 for additional details). (B) T-Ag binds to and inactivates p53, which prevents inhibition of the cell cycle or apoptosis. (C) T-Ag binds to IRS-1, a key member of the IGF signalling pathway, leading to uncoupling of IGF-1R from IRS-1, and its translocation to the nucleus. In the nucleus, IRS-1 may be important in Rad51 trafficking and homologous recombination directed DNA repair. (D) Finally, T-Ag can be oncogenic by deregulating the Wnt signalling pathway. In normal cells, β-catenin levels are regulated by a multiprotein complex of GSK3β, Axin-1, and APC, which phosphorylate β-catenin and trigger its degradation. On JCV infection, T-Ag binds to β-catenin, leading to its stabilisation, and providing a nuclear localisation signal. Once in the nucleus, stabilised β-catenin complexes with transcription factors such as TCR/LEF, which stimulates the transcription of cell cycle regulatory genes, including c-myc and cyclin D1.
JCV and genomic instability: implications for colorectal carcinogenesis
Molecular pathways responsible for genomic instability in CRC include microsatellite instability (MSI), CIN, and CpG island methylator phenotype (CIMP). Approximately 12–15% of all CRCs demonstrate MSI, a reasonably well understood process caused by inactivation of the DNA mismatch repair (MMR) system. However, the majority of CRCs acquire genomic instability by one of two other pathways. CIN is characterised by aneuploidy and loss of heterozygosity (LOH), and leads to losses of tumour suppressor genes on chromosomes 5q, 18q, and 17p. In a completely unique pathway, CIMP leads to cancer by silencing tumour suppressor genes through hypermethylation of CpG sequences in the promoters of these genes.27 The biological bases of these two critical processes are not currently understood. It has long been appreciated that CRCs are commonly aneuploid and have CIN, but the molecular mechanisms causing CIN are poorly understood, and are currently an issue of controversy and intense investigation.
CIN produces large scale chromosomal losses, eliminating millions of base pairs at once, and unlike point mutations, which can create a variety of silent or innocuous alterations at the amino acid level, deletions unambiguously inactivate genes. Colon cancers typically have multiple LOH events that occur diffusely throughout the genome. In some instances, more than 50% of assayed loci have experienced LOH.28 Losses of the APC gene have been proposed as a cause of CIN.29 However, LOH is commonly the mechanism for inactivation of APC2,28; so it would seem circular to propose that CIN is both the result of losing APC and the cause of LOH at APC. This argues for the primary occurrence of some form of genomic instability, the random generation of genomic diversity, the consequent loss of tumour suppressor genes, and the expansion of favoured clones in an evolutionary fashion. However, there is no convincing evidence for a genetic event that accounts for CIN in most, or even a large proportion of, CRCs.
We hypothesised that a transforming virus, such as JCV, which ubiquitously infects most humans and encodes the highly oncogenic T-antigen, would interact with key signalling pathways, simulate Wnt signalling, and abrogate the functions of p53 and pRb, which would provide the right milieu of cellular resources and opportunity for this virus to induce CIN in CRCs.30 Armed with the speculation that JCV might cause CIN, our laboratory first determined that JCV DNA sequences were present in human colon cancers and adjacent normal colonic tissues. In pilot studies, we found that the native supercoiled JCV was difficult to amplify by polymerase chain reaction (PCR). PCR amplification following pretreatment of viral DNA with topoisomerase I (which relaxes supercoiling) and the use of degenerate primers (anticipating microheterogeneity) allowed us to detect JCV in 89% of colon cancers, but also in 89% of normal adjacent colonic epithelia from the resected matched tissue specimens.17,31 These findings have been confirmed in another laboratory, in which JCV DNA sequences were found in 81% of colon cancers.18 Furthermore, expression of several viral proteins, including T-antigen and agnoprotein, occurs in benign adenomas and tumour specimens, but not in any normal colonic tissues, suggesting that JCV may play a role in the earliest stages of neoplastic development in the colon. Subsequently, using a semiquantitative PCR assay, we found at least 10-fold more copies of the virus in colon cancers than in the adjacent normal colon.17
Encouraged by these findings, and cautiously optimistic, we tested the hypothesis that JCV latently infects the gastrointestinal tracts of individuals without neoplasia. We obtained biopsy samples from the oesophagus, stomach, duodenum, colon, and rectum from patients undergoing routine diagnostic examinations and found that 75.8% of patients harboured JCV sequences (70.6% of the upper gastrointestinal samples and 81.2% of the colorectal samples). These data were subsequently confirmed using nested PCR, Southern blot analysis, and direct DNA sequencing analysis.11 We concluded that the gastrointestinal tract is a reservoir for JCV and proposed that ubiquitous infection of humans with this virus may be due to faecal-oral transmission during childhood. More recently, we sequenced the TCR of JCV from isolates of CRCs and adjacent normal tissues and found them to be the Mad-1 strain, which differs slightly from the archetypal strain commonly found in lymphocytes and urine.32
The APC tumour suppressor is a negative regulator of the Wnt signalling pathway in normal colonic epithelium. APC, in conjunction with AXIN and GSK-3β, forms a complex necessary for the phosphorylation and degradation of β-catenin, thereby preventing upregulation of growth controlling genes such as c-MYC and cyclin D1. Most CRCs have undergone biallelic inactivation of the APC gene, and allelic loss of one allele is a common and early event.28,33 Recent data from the laboratory of Khalili and colleagues18 have also demonstrated the ability of T-antigen to interact with β-catenin. These results suggest the interpretation that expression of T-antigen could stabilise β-catenin before loss of APC and create a state of hyperproliferation prior to actual loss of APC or other tumour suppressor genes, and allow the emergence of CIN. Taking cues from the indirect but compelling evidence for the possible role of JCV T-antigen in inducing CIN in the colon, Ricciardiello and colleagues34 tested the hypothesis that JCV T-antigen may directly induce CIN in RKO cells, which is a diploid CRC cell line with wild-type APC, p53, and β-catenin. The authors found that transfection of plasmid DNA containing the Mad-1 strain of JCV in these cells led to expression of T-antigen and viral capsid proteins. These cell lines developed CIN within seven days of transfection, providing direct experimental evidence for the ability of T-antigen to induce CIN in colonic epithelial cells.
In summary, we and others have demonstrated that most CRCs contain the DNA of JCV that encodes an oncogenic T-antigen, which is capable of interacting with key growth regulatory pathways in the colon, and has the potential to induce CIN. Current evidence indicates that JCV infection is ubiquitous and remains subclinical throughout the life of most individuals, but can cause serious disease in the setting of extreme immunosuppression. It is proposed that some factor or factors lead to activation of the virus in the colon, induction of the adenomatous phenotype, CIN, and eventually colon cancer. Finally, based on unpublished data, it appears that the virus may be too disruptive for the highly proliferative cancer in its advanced stages, and the infection may be lost in a “hit-and-run” fashion. Thus we are suggesting that JCV is involved in the initiation of colorectal neoplasia, but the instability may be selected against once an optimal reorganisation of the neoplastic genome has been achieved.
INFLAMMATION AND GASTROINTESTINAL CANCER
Association between inflammation and gastrointestinal cancer
Over 150 years ago, the German pathologist Virchow described a link between inflammation and cancer.35 A number of observations strongly suggest that cancer is a common consequence of chronic inflammation in the gut. Gastrointestinal cancers are more prevalent in disease states that produce a chronically inflamed epithelium (table 1). The emergence of multifocal cancers in inflamed mucosae adds to the speculation that inflammation is mechanistically involved in carcinogenesis.
Association between chronic inflammation and cancer in the gastrointestinal tract
The best data on inflammation driven carcinogenesis come from studies in ulcerative colitis. Ulcerative colitis affects approximately 0.3% of Western populations and typically starts in the second or third decade of life. The pathogenesis of ulcerative colitis is only partially understood and involves autoimmunity, genetic predisposition (for example, the IBD2 locus on chromosome 12q), and environmental triggers. The first case of CRC in ulcerative colitis was described by Crohn and Rosenberg in 1925.36 Since then, epidemiological evidence has clarified that patients with long term ulcerative colitis have an increased risk of developing CRC. The magnitude of this risk has been difficult to estimate but was considered very high (increased up to 50-fold) in early studies.37–44 Later studies based on population cohorts gave rise to reduced estimates.45,46 The risk increases with disease extent (19-fold in pancolitis),47 severity of inflammation,48 young age at onset,46 family history of colorectal cancer,49 presence of primary sclerosing cholangitis,50,51 and presence of backwash ileitis.52
Several genetic and epigenetic changes have been described in ulcerative colitis that might be responsible for colon carcinogenesis, and these differ in frequency and timing compared with sporadic cancer.53,54 One can conceptually categorise CRCs by the mutational signature found in the tumour tissue (that is, CIN, MSI, or CIMP), as described above, but colorectal carcinogenesis in ulcerative colitis may be unique. Early events in colitis associated cancers often involve MSI and p53 mutations, which are present in non-dysplastic chronically inflamed tissue. These features may serve as a starting point to unravel the mystery of inflammation associated CRC.
Mechanisms involved in inflammation associated cancer
In aerobic cells, reactive oxygen species (ROS) are generated as a byproduct of normal mitochondrial activity. Major endogenous sources are metabolic processes, primarily oxidative metabolism in the mitochondria, and pathological processes such as inflammation. Exogenous sources of ROS include exposure to ionising radiation or radiomimetic chemicals, and ROS produced by neighbouring cells. Toxic ROS include oxygen species such as the superoxide anion radical (•O2−), hydrogen peroxide (H2O2), hydroxyl radical (•OH), as well as nitric oxide (NO). ROS can cause severe damage to cellular macromolecules, especially DNA.
DNA damage is detected in the nucleus by various protein complexes, so called sensor proteins, which initiate the cellular response (fig 3). These sensor proteins activate repair pathways that include transducer, signal, and effector proteins. DNA damage is processed through specific repair pathways, including base excision repair (BER), DNA MMR, and nucleotide excision repair (NER), depending on the type of damage involved. Cell cycle checkpoints function as protective barriers against cytotoxicity and carcinogenesis induced by ROS by providing damaged cells additional time to repair DNA before replication or mitosis (fig 4). When damage overwhelms the ability to repair, checkpoint signalling may cause cells to undergo apoptosis or enter an irreversible G0 state.55 It is critical to appreciate that as cancer develops in the setting of oxidative stress, ROS not only damage DNA, they also inactivate some of these intracellular protective mechanisms, a deadly combination (fig 5).

Cellular response to cytotoxic stress. The MRN complex, whose core contains the proteins Mre11, Rad50, and Nbs1, functions as the sensor of DNA double strand breaks, and is required for recruiting the transducer proteins for the DNA damage response. Sensor proteins recognise either the lesions themselves or chromatin alterations that follow the DNA damage, and transmit the information to transducer proteins such as the ataxia-telangiectasia mutated kinase (ATM) and the ATM and Rad related kinase (ATR), which control the damage response through phosphorylation of effector proteins. Individuals with ataxia-telangiectasia (AT), whose manifestations include genome instability and a predisposition to cancer, have mutations in the gene coding for the protein kinase ATM. Downstream of the transducer proteins are targets that control various cellular processes, such as DNA repair, cell cycle progression, gene transcription, protein synthesis and degradation, and apoptosis. Targets include the protein kinases Chk1 and Chk2, which are involved in activation of the G2/M cell cycle checkpoint.
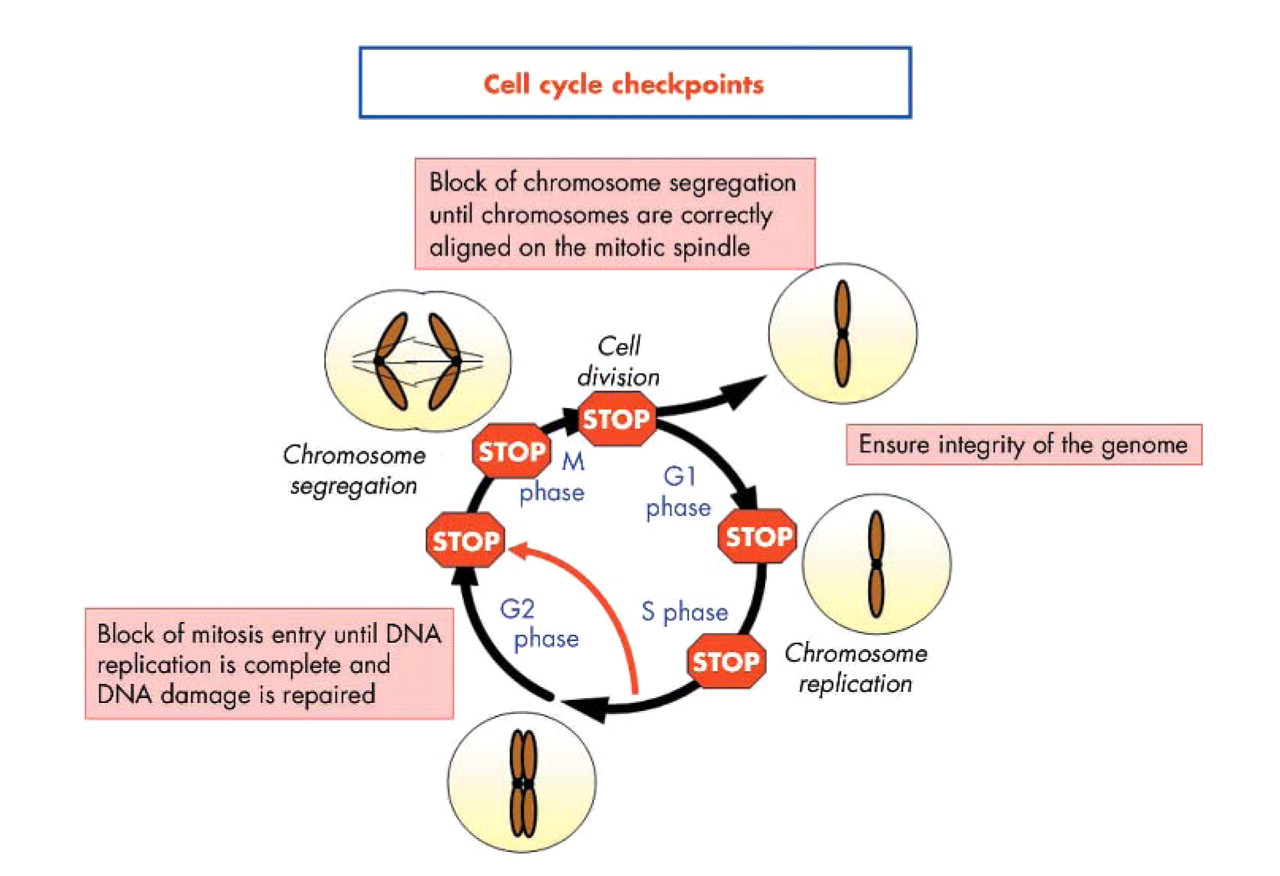
Cell cycle checkpoints. In a dividing cell, the transitions between the phases of the cell cycle (G1 to S to G2 to mitosis) are regulated by the sequential activation of molecular engines called cyclin dependent kinases (cdks). These kinases regulate the precise moments at which each step—such as DNA synthesis, protein synthesis, and cell division—will occur. In response to genotoxic stress such as DNA damage, oxidative stress, ionising radiation, and methylating agents, cellular checkpoints may become activated and block the cell in one of the various phases of the cell cycle. The G1/S checkpoint ensures that only undamaged or repaired DNA undergoes replication. The G2/M checkpoint provides sufficient time for correction of post-replication errors before mitosis. In addition to these checkpoints, intra-S arrest may be activated in response to aberrations that occur during DNA replication, which arrests the cell cycle until the problem is solved. Finally, if the chromosomes are not properly aligned on the mitotic spindle, the M phase or spindle checkpoint is triggered to prevent the cell from dividing, which prevents mitotic catastrophes. If DNA damage is overwhelming and cannot be repaired, apoptosis may ensue.

The paradigm of inflammation and cancer. Oxidative stress causes various forms of DNA damage that are typically corrected by the appropriate repair pathway. Oxidative stress also downregulates DNA repair activity, which permits the excessive accumulation of mutations, a curiously maladaptive combination.
The tumour suppressor, p53
Mutations of the p53 gene are the most common genetic alterations found in human cancers, and these play crucial roles in tumour development and progression. In sporadic CRC, p53 protein stabilisation can be caused by p53 mutations, overexpression, or reduced degradation, and this occurs late in carcinogenesis. However, p53 stabilisation—usually attributed to mutation—is found in early dysplastic lesions in ulcerative colitis, and even in chronically inflamed mucosa without dysplasia.56 Similarly, p53 mutations have been detected in inflamed non-neoplastic synovia from patients with rheumatoid arthritis.57 LOH of p53 is also common in ulcerative colitis associated dysplasia or carcinoma. The p53 gene encodes a protein that functions like a transcription factor, and the biological functions of p53, which becomes activated in the nucleus in response to genotoxic stress, are manifested through the activities of its downstream genes (for reviews see Hupp and colleagues,58 Woods and Lane,59 and Bartek and Lukas,60 and also fig 6).

The p53 pathway. The p53 tumour suppressor protein plays a central role in the cellular response to a range of environmental and intracellular stresses, including agents which cause DNA strand breaks, ultraviolet radiation, and oxidative stress. These signals are transduced because p53 is a transcription factor that binds to the promoters of genes involved in the DNA damage response programme. The biological outcomes of p53 induction include growth arrest, DNA repair, and apoptosis. Among p53 responsive genes are those involved in growth arrest such as the CDK inhibitor protein p21WAF1, which mediates G1/S arrest by blocking cyclin E-Cdk2 mediated phosphorylation of pRb. Many proapoptotic genes are also stimulated by p53, particularly those involved in the mitochondrial pathway of apoptosis such as BAX, and APAF1. Under normal circumstances, p53 is tightly regulated through its interaction with MDM2, a negative regulatory partner. MDM2 is an E3 ubiquitin ligase, which mediates both ubiquitination and proteasome dependent degradation of p53. Finally, p53 regulates GADD45a, which is involved in DNA repair. The GADD45a protein interacts with Cdc2 protein kinase, proliferating cell nuclear antigen (PCNA), and p21WAF1 protein, indicating that GADD45a may play a role in cell cycle checkpoint control, DNA repair, and signal transduction. GADD45a appears to be an important component in the cellular defence network required for maintenance of genomic stability.
Oxidative stress has been found to initiate checkpoint arrest in several eukaryotic cell types.61 Tumour suppressor p53 plays a vital role in the G1 checkpoint function and its loss results in near complete ablation of that checkpoint. Cells that have incurred DNA double strand breaks during the G1 phase activate and stabilise p53 primarily via an ATM dependent pathway.62 ATM regulates p53 accumulation by indirect pathways involving Chk2 phosphorylation of p53 and MDM2, thus blocking the interaction between MDM2 and p53. Exposure to oxidative stress enhances p53 and increases intracellular protein levels.63 Moreover, p53 is post-translationally modified and accumulates in response to the free radical NO•. The latter can also increase BER enzymes AAG and APE1 level and activity.64
The majority of mutations in the p53 gene involve exons 7 and 8. Transition mutations in p53 codons 247 and 248 are common in the inflamed mucosa of ulcerative colitits patients.56 Analysing the mutational signatures provides clues to the links between oxidative stress and cancer. Base transitions are typical consequences of oxidative DNA damage. The intermediates of lipid peroxidation and alkylating agents can induce G to A mutations; C to T transitions may be due to the formation of 5-hydroxycytidine, or to the deamination of 5-methylcytosine at CpG sites.65 The early appearance of p53 alterations makes this an attractive potential marker to screen for ulcerative colitits associated malignancy and in the assessment of cancer risk.
The tumour promoting gene nuclear factor κB (NFκB)
The NFκB family includes sequence specific transcription factors and consists of a number of related proteins that bind a common sequence motif known as the κB site. Members of the NFκB family exist in unstimulated cells as homo- or heterodimers bound to the inhibitory family of proteins called IκB. Binding to IκB prevents the complex from translocating to the nucleus, thereby maintaining NFκB in an inactive state. NFκB signalling occurs through either a classical or an alternative pathway. In the classical pathway, NFκB activation may occur in response to stimulation by tumour necrosis factor α, interleukin 1, bacterial lipopolysaccharide, viral double stranded RNA, ionising radiation, etc. Stimulation of this signalling pathway leads to activation of the β subunit of the IκB kinase (IKK) complex, which then phosphorylates IκB proteins. Phosphorylation of IκB leads to ubiquitin dependent degradation of IκBs, which allows NFκB dimers to translocate to the nucleus. NFκB proteins are transcription factors that target expression of inflammatory and immunoregulatory genes, antiapoptotic genes (such as members of the Bcl2 family such as cIAP1/2 and Bcl-XL), cell cycle control genes, and genes that code for negative regulators of NFκB.66
Activation of NFκB by mutations, chromosomal rearrangements, or chronic inflammation plays an important role in tumour development by cultivating mutations (tumour promotion) and by preventing cells with mutations from undergoing apoptosis.67 In most types of cells, this leads to accelerated cell cycle progression, an antagonised interaction with p53, elevated resistance to radiation and chemotherapy, as well as increased invasive growth and metastasis. Therefore, NFκB plays a key role in promoting inflammation associated cancer, and is a potential target for cancer prevention in chronic inflammatory diseases. Also, tumours that have constitutive NFκB activity usually have increased resistance to chemotherapy.68 In turn, many chemotherapeutic agents induce NFκB activity, thereby increasing drug resistance in tumour cells. Moreover, NFκB interferes with p53 mediated genome surveillance mechanisms by upregulating antiapoptotic genes and downregulating p53 levels. This ultimately leads to the survival of cells with damaged or mutated DNA that are normally eliminated by p53 mediated apoptosis.
In addition to its tumour promoting role, NFκB may also participate in tumour initiation. Activation of NFκB in macrophages and neutrophils can promote the production of ROS through induction of NO synthase. ROS produced in these cells cause DNA damage and carcinogenic lesions in surrounding cells.66 Using ATM knockout cells, and cells from patients with ataxia telangectasia (AT), it has been shown that ATM is required for NFκB activation following ionising radiation.69
Activation of NFκB in the gastrointestinal tract leads to induction of proinflammatory cytokines which maintain inflammation. Intestinal NFκB activation has been found in patients with Crohn’s disease and ulcerative colitis. Using a colitis associated cancer model, Greten and colleagues70 showed that deletion of IKKβ from intestinal epithelial cells led to a decrease in tumour incidence (primarily due to increased epithelial apoptosis during tumour promotion) without decreasing the degree of inflammation.
Several natural compounds and synthetic drugs that are able to inhibit the IKK/NF-κB activation pathway have been shown to either prevent cancer or to inhibit cell growth in animal models.71 Aspirin and non-steroidal anti-inflammatory drugs (NSAIDs) that reduce the incidence of colorectal cancer in animal models and the risk of cancer throughout the gut in humans may exert some of their function by inhibiting IKK dependent activation of NFκB. However, the action of these inhibitors is not solely directed towards the NFκB pathway.
DNA repair pathways: BER, NER, and MMR
DNA repair pathways are complex activities that involve excision of the damaged base, restoration of the original DNA sequence by a DNA polymerase that uses the undamaged strand as its template, and sealing of the remaining break in the double helix by DNA ligase (for review see Bartek and colleagues62). The major DNA repair mechanisms, and the types of damage repaired by each, are illustrated in fig 7. The base excision repair (BER) pathway includes enzymes that recognise a specific type of altered base in DNA (such as 8-oxo-guanidines that result from nucleotide oxidation) and catalyse its removal. Another major repair pathway is nucleotide excision repair (NER), which can repair the damage caused by “bulky adducts”, as these will change the structure of the DNA double helix. However, members of the MMR pathway are commonly required to complete the DNA repair.72

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DNA repair mechanisms. This diagram illustrates the different DNA repair cellular pathways triggered by diverse damaging agents, and the repair mechanisms required for each. SSB, DNA single strand breaks; DSB, DNA double strand breaks; BER, base excision repair system; MMR, DNA mismatch repair; NER, nucleotide excision repair.
Some types of DNA damage require unique repair strategies. Double stranded DNA breaks can be caused by ionising radiation, oxidising agents, replication errors, and certain metabolic byproducts in the cell. Two mechanisms have evolved to repair this damage: non-homologous end joining (NHEJ) and homologous recombination.73 The latter requires factors that recognise areas of DNA sequence matching between the two chromosomes and physically bring them together. Although NHEJ may induce changes in the DNA sequence at the site of breakage, so little of the mammalian genome codes for proteins that this mechanism is an acceptable solution to the problem of keeping chromosomes intact, even at the risk of inducing mutations.
Cells may respond to stress by elevating levels of DNA repair enzymes, as an emergency response to DNA damage.64 Induction of these additional DNA repair enzymes increases cell survival after DNA damage but also transiently elevates the mutation rate by increasing the number of errors made while copying DNA sequences. Such errors may be caused by low fidelity DNA polymerases that are used for DNA synthesis during MMR74,75 Minor DNA polymerases are specifically called into play to copy unrepaired lesions in the DNA template. These enzymes can recognise specific types of DNA damage and add nucleotides to restore the initial sequence.
Approximately 15% of sporadic CRCs have a mutational signature called microsatellite instability (MSI). The MSI pathway is a consequence of losing the activities of enzymes that repair DNA base pair mismatches that occur during the normal process of DNA replication. These errors preferentially target genes such as TGF-βRII or BAX, because exons in these genes contain repeated sequences of mononucleotides (that is, An, Gn, etc.) that are intrinsically unstable and prone to be mutated during DNA replication. If the repetitive sequence is more than six or seven consecutive nucleotides, it is highly dependent on DNA MMR activity for faithful replication, and will be mutated once in every 102–103 replications in the absence of this repair system. Germline mutations in the MMR genes are responsible for Lynch syndrome, or hereditary non-polyposis colorectal cancer,76 but this does not cause the MSI that is frequently found in ulcerative colitis. Therefore, something other than germline mutations cause defective MMR in the chronically inflamed colon.
Chang et al found that H2O2 inactivates the DNA MMR system, apparently by damaging the enzymes at the protein level.77 More direct evidence for the role of oxidative stress in colon carcinogenesis via MMR inactivation has come from experiments that induce frameshift mutations in a reporter gene after exposure of cells to hydrogen peroxide.78 This effect occurs in both MMR proficient and MMR defective cell lines. These findings suggest that oxidative stress may temporarily “relax” the MMR pathway, which permits MSI in the absence of inactivating germline mutations in the MMR genes. This may contribute to the MSI-low (MSI-L) phenotype (with a lower frequency of microsatellite mutations79), and in this instance, the microsatellite mutations are found in dinucleotide repeats.80 Dinucleotide repeats are very rarely found within exons but are very common in non-coding DNA. Importantly, this mutational signature is present in non-neoplastic80 as well as in neoplastic mucosa of ulcerative colitis patients.81 In a subset of patients with MSI-high (MSI-H) CRCs, the mechanism is reported to be attributable to hMLH1 promoter hypermethylation, which suggests that the process is complex and may involve multiple mechanisms.82 As mentioned above, BER activity is significantly increased in non-cancerous colons of ulcerative colitis patients, and MSI correlates with an adaptive imbalance in DNA repair activities.64 Furthermore, the imbalance in BER enzymes increases the generation of spontaneous mutations. Interestingly, the BER enzyme APE-1 promoter contains the consensus sequence for binding NFκB, suggesting additional crosstalk between the inflammatory response and the repair response.
Anti-inflammatory drugs and colorectal cancer prevention
Based on the issues mentioned above, efforts have been made to interfere with carcinogenesis by manipulating inflammatory pathways. Firstly, there is strong evidence for an inverse relationship between aspirin or NSAID consumption and CRC incidence and mortality. It was initially assumed that the mechanism for the anticancer effect of NSAIDs was reduced prostaglandin synthesis by inhibiting cyclooxygenase (COX) activity.83 However, several COX independent mechanisms of NSAID related chemoprevention have been found, including induction of apoptosis by regulating p38 kinase and MMR protein activities,84,85 transcriptional activation through interactions with PPARγ,86 and inhibition of transcription by binding to IKKβ.87
Two large randomised trials have confirmed the beneficial effects of aspirin in reducing the formation of colorectal adenomatous polyps.88,89 Interestingly, both studies reported significant effects of aspirin in preventing colorectal adenomas, although one study used a much lower dose of the drug (81 mg) compared with the other trial (325 mg). Because of the toxicity of aspirin and related drugs, selective COX-2 inhibitors (coxibs) were developed in an attempt to improve safety profiles. Although the efficacy of the coxibs is still a question, the recently reported toxicities of this class of drugs makes it unlikely that these will emerge as clinically useful chemopreventive agents.90,91 Consequently, another potentially preventive agent is nitric oxide releasing aspirin which may be an interesting candidate for future prevention trials
Although structurally quite similar to aspirin, mesalamine (5-aminosalicylic acid) has different biological properties. It is only a weak inhibitor of COX-2 and does not prevent recurrence of sporadic polyps.92 However, several studies have suggested that the long term use of 5-ASA in ulcerative colitis patients may significantly reduce the risk of development of colorectal cancer (summarised in Eaden93). Some effects may be due to its oxygen scavenging properties and other effects to its general efficacy as a therapeutic agent in ulcerative colitis. It has been more recently reported that mitotic arrest in response to treatment with 5-ASA may be independent of its anti-inflammatory properties.94 Interestingly 5-ASA, but not aspirin, improves replication fidelity.95 This compound seems to act differently than aspirin, and might be clinically useful in preventing different types of CRC such as those due to chronic inflammation or low replication fidelity (as in Lynch syndrome).
SUMMARY AND CLINICAL IMPLICATIONS
Our appreciation of the basic mechanisms of carcinogenesis has moved from a description of the mutated gene targets to an understanding of the mechanisms by which these mutations are generated, and the exogenous influences that modify the rates of mutation. One can only understand CRC in the context of the processes that lead to the evolution of neoplasia. Analysis of the “mutational signatures” in neoplastic tissue permits classification of CRCs, and raises the perplexing possibility that CRC might be at least three different diseases. The possible role of the common polyomavirus, JCV, in the genesis of CRC with chromosomal instability raises new possibilities for our understanding of this disease, and possible novel opportunities for prevention. Inflammation is clearly associated with an increased risk of mucosal neoplasia throughout the gut but the mechanisms by which this occurs are just beginning to be clarified. This appears to be a complex process, and preventive strategies may depend on the clinical context. What is appropriate for the prevention of sporadic cancers may be inappropriate for the prevention of colitis associated neoplasia. The transition of cancer research from the realm of descriptive pathology to molecular biology has been revolutionary, and will continue to modify our understanding of gastrointestinal cancer.
Acknowledgments
Supported in part by grants from the National Cancer Institute to CRB (R01 CA72851 and R01 CA98572), the Austrian Science Fund to CG (P15314 and P17943-B13) and MGL (M874-B14), and from funds of the Austrian National Bank to CG (ONB 10.543).
REFERENCES
Footnotes
-
Conflict of interest: None declared.