Article Text

Abstract
Background and aim Thymus-derived regulatory T cells (Tregs) mediate dominant peripheral tolerance and treat experimental colitis. Tregs can be expanded from patient blood and were safely used in recent phase 1 studies in graft versus host disease and type 1 diabetes. Treg cell therapy is also conceptually attractive for Crohn's disease (CD). However, barriers exist to this approach. The stability of Tregs expanded from Crohn's blood is unknown. The potential for adoptively transferred Tregs to express interleukin-17 and exacerbate Crohn's lesions is of concern. Mucosal T cells are resistant to Treg-mediated suppression in active CD. The capacity for expanded Tregs to home to gut and lymphoid tissue is unknown.
Methods To define the optimum population for Treg cell therapy in CD, CD4+CD25+CD127loCD45RA+ and CD4+CD25+CD127loCD45RA− Treg subsets were isolated from patients’ blood and expanded in vitro using a workflow that can be readily transferred to a good manufacturing practice background.
Results Tregs can be expanded from the blood of patients with CD to potential target dose within 22–24 days. Expanded CD45RA+ Tregs have an epigenetically stable FOXP3 locus and do not convert to a Th17 phenotype in vitro, in contrast to CD45RA− Tregs. CD45RA+ Tregs highly express α4β7 integrin, CD62L and CC motif receptor 7 (CCR7). CD45RA+ Tregs also home to human small bowel in a C.B-17 severe combined immune deficiency (SCID) xenotransplant model. Importantly, in vitro expansion enhances the suppressive ability of CD45RA+ Tregs. These cells also suppress activation of lamina propria and mesenteric lymph node lymphocytes isolated from inflamed Crohn's mucosa.
Conclusions CD4+CD25+CD127loCD45RA+ Tregs may be the most appropriate population from which to expand Tregs for autologous Treg therapy for CD, paving the way for future clinical trials.
- IBD CLINICAL
- IMMUNOTHERAPY
- INTESTINAL T CELLS
- IBD
- IBD BASIC RESEARCH
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 3.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Thymically derived regulatory T cells (Tregs) can modulate effector immune responses and, when expanded in vitro, have recently shown promise for graft versus host disease and type 1 diabetes in humans, leading to interest in this therapeutic approach for Crohn's disease.
Barriers to autologous Treg therapy in Crohn's include the requirement for in vitro expansion to a target dose, potential Treg plasticity to pathogenic interleukin-17+ cells, uncertain homing to mucosal tissue and effector T cell resistance to Treg-mediated suppression in inflamed Crohn's mucosa.
Initial enrichment on the basis of CD45RA+ expression can improve the phenotypic stability of an expanded Treg population obtained from healthy control blood, but the value of this approach in Crohn's disease is unknown.
What are the new findings?
We show that it is technically feasible to expand functional Tregs to numbers consistent with a target dose from the blood of patients with Crohn's disease.
In vitro expansion enhances the in vitro suppressive activity of these cells. Expanded Tregs suppress activation of lamina propria and mesenteric lymph node lymphocytes isolated from inflamed Crohn's mucosa.
In contrast to Tregs expanded from CD45RA− precursors, expanded CD45RA+ Tregs have epigenetically stable FOXP3 expression and are resistant to Th17 conversion.
Expanded CD45RA+ Tregs also express α4β7 integrin, CD62L and CCR7, and home to human small bowel in a SCID mouse bearing subcutaneously implanted human intestine.
Introduction
Thymically derived FOXP3+ regulatory T cells (Tregs) are key mediators of peripheral tolerance and are likely to have a role in preventing inappropriate mucosal inflammation in response to bacterial, and other, luminal antigens. In mice, Treg depletion impairs oral tolerance.1 Adoptively transferred Tregs prevent the onset of colitis or treat established colitis in a number of murine models.2–7 FOXP3 mutations lead to multisystem autoimmunity with enteropathy in mice and humans.8 ,9 Disruption of other key molecules implicated in Treg function, such as transforming growth factor (TGF)-β, Cytotoxic T Lymphocyte-Associated (CTLA)-4, interleukin (IL)-10R subunits, IL-2 or its receptor subunits, is associated with autoimmunity and intestinal inflammation.10
Human peripheral blood (PB) or umbilical cord blood Tregs can be expanded in vitro through T cell receptor (TCR) stimulation in the presence of IL-2.11–26 In vitro expanded human Tregs prevent transplant rejection,27 ,28 transplant arteriosclerosis29 and graft versus host disease (GvHD)21 ,30 in humanised mice. Promisingly, recent phase 1 clinical trials have shown Treg cell therapy to be safe in patients with GvHD12 ,24 and type 1 diabetes.18 Additional phase 1 studies have started in renal (the ONE study) and liver transplantation (ThRIL study).19 ,31
Lamina propria (LP) Tregs are increased in the mucosa of patients with active Crohn's disease (CD) and decreased in blood, compared with healthy controls.32–34 LP Tregs obtained from inflamed CD mucosa suppress proliferation of conventional CD4+CD25lo/int T cells (Tcon) obtained from blood but not LP Tcons,35 suggesting that mucosal Tcons in active CD may be resistant to Treg-mediated suppression. LP Tcons from CD mucosa overexpress Smad7, an inhibitor of TGF-β signalling, which confers resistance to Treg-mediated suppression.35 ,36 Activated Tcons also have an effector-memory phenotype, conferring relative resistance to Treg-mediated suppression.37 However, Tregs expanded in vitro in the presence of rapamycin from the PB of patients with end-stage renal failure (ESRF), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS) and asthma are more suppressive than freshly isolated Tregs obtained from the same donor.26 ,38 If it can be shown that in vitro expansion enhances the suppressive ability of CD PB Tregs and that these expanded cells suppress mucosal inflammation, parenteral therapy with autologous in vitro expanded Tregs generated from CD PB would become a conceptually attractive approach to induce remission in active CD.
IL-17 contributes to mucosal homoeostasis but has also been implicated in the pathogenesis of CD. Tregs isolated from healthy donor PB or tonsils can be induced to express IL-17 and the Th17 transcription factor RORC when activated in vitro in the presence of IL-1, IL-2, IL-21 and IL-23.39–42 Although major sources of IL-17 in the gut include Tcons and γδ T cells, a proportion of Tregs obtained from inflamed CD mucosa co-express FOXP3 and IL-17.43 Th1 Treg plasticity has also been described in vitro and in vivo.44 ,45 In humans, phenotypically distinct Treg populations can be delineated on the basis of CD45RA expression.17 ,46 ‘Resting’ CD4+CD25hiCD127loCD45RA+ Tregs (rTregs) are resistant to induction of IL-17 and interferon (IFN)-γ in vitro.46 In contrast, ‘activated’ CD4+CD25hiCD127loCD45RA− Tregs (aTregs) can be induced to express IL-17 and IFN-γ in vitro.46 Similarly, Tregs expanded from healthy donor CD45RA+ Tregs (in the absence of rapamycin) do not contain cytokine producers and are highly suppressive, while Tregs expanded from CD45RA− Tregs express proinflammatory cytokines and lose FOXP3 expression with repetitive stimulation in vitro.17 ,47 Tregs expanded from healthy control CD45RA− precursors (but not CD45RA+ precursors) also have stimulation-induced demethylation of RORC, which may be permissive for IL-17 expression.48 Furthermore, imprinting α4β7 integrin expression on in vitro expanded Tregs by supplementing culture with all-trans retinoic acid (ATRA) results in high IL-17 expression.21 Even though IL-17+ Tregs isolated from human blood and tonsil retain their suppressive ability in vitro,39–41 the potential for adoptively transferred Tregs to exacerbate inflammation in CD lesions through the production of proinflammatory cytokines is of significant concern.
Using cell enrichment strategies achievable with currently available good manufacturing practice (GMP) technologies, we show that initial enrichment on the basis of CD45RA+ expression is required to generate a homogenous and epigenetically stable Treg population following expansion, in the presence of rapamycin, from the PB of patients with CD. These cells are resistant to Th17 plasticity, express lymphoid and gut homing markers, and home to human gut following adoptive transfer to a SCID mouse bearing subcutaneously implanted human small bowel (SB). In vitro expansion also enhances the suppressive ability of these cells, licensing them to suppress activation of LP and mesenteric lymph node (MLN) Tcons obtained from inflamed CD resection specimens. These data suggest that CD PB CD4+CD25hiCD127loCD45RA+ cells may be the most appropriate population from which to expand Tregs in vitro for forthcoming clinical trials of autologous Treg cell therapy in CD.
Materials and methods
Patient samples
Following Institutional Review Board (IRB) approval (SE London REC 2; 10/H0804/65 and East London REC 2 (10/H0704/74)), patients with CD attending Guy's & St Thomas’ National Health Service (NHS) Foundation Trust and Bart’s Health NHS Trust were invited to donate blood and/or resected tissue. Prospective written consent was obtained. Demographic details are shown in table 1.
Demographic details of study patients
Treg enrichment and sorting
Online supplementary figure S1 illustrates the experimental design. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation over lymphocyte separation medium (LSM) 1077 and CD4+ lymphocytes enriched to >95% by positive magnetic activated cell separation (MACS) selection (Miltenyi, Bergisch-Gladbach, Germany). Lymphocytes were labelled using the ‘Human Regulatory T Cell Sorting Kit’ (BD Biosciences, San Diego, California, USA), as described previously,25 and sorted to CD4+CD25hiCD127loCD45RA+ and CD4+CD25hiCD127loCD45RA− Treg subsets, and autologous CD4+CD25− Tcons on a FACSAria (BD; see online supplementary figure S2A–D). Median (IQR) postsort purity was 86.5% (80.8–91.6%; n=13) for CD4+CD25hiCD127loCD45RA+ Tregs (CD45RA+ Tregs) and 92.7% (87.7–94.9%; n=13) for CD4+CD25hiCD127loCD45RA− Tregs (CD45RA− Tregs). Autologous Tcons were stored at −80°C.
In vitro generation of Treg lines
Precursor Treg populations were expanded in vitro as described previously21 ,25 and described in detail in online supplemental methods.
Cell surface and intracellular stains
Fluorochrome-conjugated antibodies, buffers and experimental technique are listed in online supplemental methods.
Assessment of the in vitro suppressive ability of putative Tregs
Assays to determine Treg function in vitro were performed as described previously,25 ,49 and described in detail in the online supplemental methods.
rtPCR
Following total RNA extraction from Trisure (Bioline, London, UK), cDNA was synthesised using the RevertAid First Strand cDNA Synthesis Kit and multiplex rtPCR performed in duplicate using the Maxima Probe/ROX qPCR Master Mix (both Thermo Fischer Scientific) on a BioRad C1000 Thermal Cycler. Primers are listed in online supplemental methods.
Estimation of cytokine concentrations
Cytokine concentrations were estimated in culture supernatants using the Cytometric Bead Array (CBA) Human Th1/Th2/Th17 Cytokine Kit (BD) or sandwich ELISAs (R&D), as indicated.
Assessment of IL-17 production under proinflammatory conditions
In vitro generated Tregs were activated with anti-CD3/anti-CD28 beads at a 1:1 ratio and cultured at 106 cells/mL in complete Roswell Park Memorial Institute (RPMI) for 5 days at 37°C/5% CO2, supplemented with the following cytokine cocktails, as previously described:21 ,23 ,39 (A) IL-2 (10 IU/mL, Proleukin); (B) IL-2, IL-1 (10 ng/mL), IL-6 (4 ng/mL) and TGF-β (5 ng/mL); (C) IL-2, IL-21 (25 ng/mL), IL-23 (25 ng/mL) and TGF-β (all R&D Systems). Supernatant IL-17 concentrations were measured by ELISA.
Assessment of FOXP3 promoter demethylation
Genomic DNA was isolated using a ‘DNeasy kit’ (Qiagen, Manchester, UK). Bisulfite conversion and assessment of the methylation status of the FOXP3 Treg-specific demethylated region (TSDR) was performed by Epiontis.50 ,51 The genomic locations of FOXP3 and GAPDH CpG-rich regions probed have been reported.51
Isolation of LP mononuclear cells and MLN mononuclear cells
LP mononuclear cells (LPMCs) and MLN mononuclear cells (MLNMCs) were isolated as described previously52 and listed in online supplemental methods.
C.B-17 SCID mouse human intestinal xenotransplant model
Experimental design is illustrated in figure 3C. The C.B-17 SCID mouse human intestinal xenotransplant model has been described previously53 ,54 and is described in detail in online supplemental methods. IRB and IACUC approvals were obtained prospectively (Ethics Committee for Animal Experimentation, Hebrew University of Jerusalem; MD-11-12692-4 and the Helsinki Committee of the Hadassah University Hospital; 81-23/04/04). Techniques for the detection of adoptively transferred Tregs are also described in detail in online supplemental methods.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 5 (GraphPad Software Inc, La Jolla, California, USA) and the methods used are described in detail in the online supplemental methods.
Results
Tregs can be expanded from the blood of patient with CD using GMP-compatible protocols
Hoffmann et al17 showed that initial Treg enrichment on the basis of CD45RA+ expression was required to expand homogenous and stable Treg lines from healthy donors in the absence of supplemental rapamycin. Rapamycin prevents the outgrowth of contaminating Tcons in Treg cultures, and may make the requirements for the starting population less stringent.11 ,13 ,15 ,21 ,23 ,55 However, the optimum precursor population from which to expand a homogenous, suppressive and epigenetically stable Treg population from CD PB is currently unknown. In previous studies, we accomplished in vitro expansion of in vitro suppressive Tregs from healthy controls21 and renal transplant candidates.26 We sought to determine if Tregs could be expanded in vitro from the blood of patients with CD.
Freshly isolated CD4+ lymphocytes from 13 patients with CD were fluorescence-activated cell sorting (FACS)-sorted into CD4+CD25hiCD127loCD45RA+ (median (IQR) of 2200 cells/mL PB (860–4400)) and CD4+CD25hiCD127loCD45RA− subsets (3700 cells/mL (2000–4500)), then expanded in vitro in the presence of high-dose IL-2, rapamycin and anti-CD3/anti-CD28 beads. Active disease, evidenced by a Harvey Bradshaw Index >5 (n=4) or elevated C reactive protein (n=1), was not associated with a significantly reduced yield (see online supplementary figure S2E). Donor clinical characteristics are given in table 1.
Every CD45RA+ Treg line proliferated, to a median (IQR) of 175-fold (66–531; n=13) at D24 (figure 1A). In contrast, 3 of 13 (23%) CD45RA− Treg lines did not proliferate and were discontinued. CD45RA− Tregs expanded 130-fold (8–209; n=10). Expanded Tregs were exclusively CD4+ lymphocytes. Expression of CD25 and FOXP3 was comparable in D24 CD45RA+ and CD45RA− Tregs (figure 1B), but a greater proportion of CD45RA+ Tregs maintained a CD4+CD25hiCD127loFOXP3+ phenotype (p=0.037; figure 1C).

Expansion, phenotype and potency of in vitro expanded Tregs. (A) Cumulative fold expansion of Treg lines at days 12 and 24 of culture, grouped according to CD4+CD25hiCD127loCD45RA+ or CD4+CD25hiCD127loCD45RA− precursors; n=13 each, bar: median. (B) Representative FACS plots gated on live events showing CD25 and FOXP3 expression at D24. (C) Proportion of Tregs with a CD4+CD25hiCD127lo Treg phenotype at D24. (D) Representative plots from a proliferation assay, illustrating dose-dependent suppression of Tcon proliferation by CD45RA+ Tregs. Proliferation CTV-labelled autologous CD4+CD25− Tcons alone (filled) or with Tregs at various Tcon:Treg ratios (bold line) is shown. (E) D24 Treg-mediated suppression of Tcon proliferation. Cumulative data showing mean±SEM suppression seen at each Tcon:Treg ratio. Pooled data from 29 Treg lines. Comparisons between suppression seen in study conditions and mean non-specific suppression seen in ‘2X’ control condition (dotted line) are shown. *p<0.05,***p<0.001 and ****p<0.0001. Tregs, thymus-derived regulatory T cells; FACS, fluorescence-activated cell sorting; Tcons, conventional CD4+CD25lo/int T cells; CTV, Cell Trace Violet; NS, not significant.
Proliferation assays were performed to determine if in vitro expanded Tregs retained the ability to suppress proliferation of autologous CD4+CD25− Tcons. CD45RA+ and CD45RA− Tregs suppressed Tcon proliferation to an equivalent degree (figure 1D–E), demonstrating specific suppression (vs the 2X cell density control) above an 8:1 Tcon:Treg ratio. CD45RA+ and CD45RA− Tregs reduced IL-2 expression in 96 h co-culture supernatants (see online supplementary figure S3A). CD45RA+ Tregs also suppressed IFN-γ expression in 96 h co-culture supernatants (see online supplementary figure S3B).
In vitro expanded CD45RA+ Tregs are resistant to IL-17 induction and stably express FOXP3
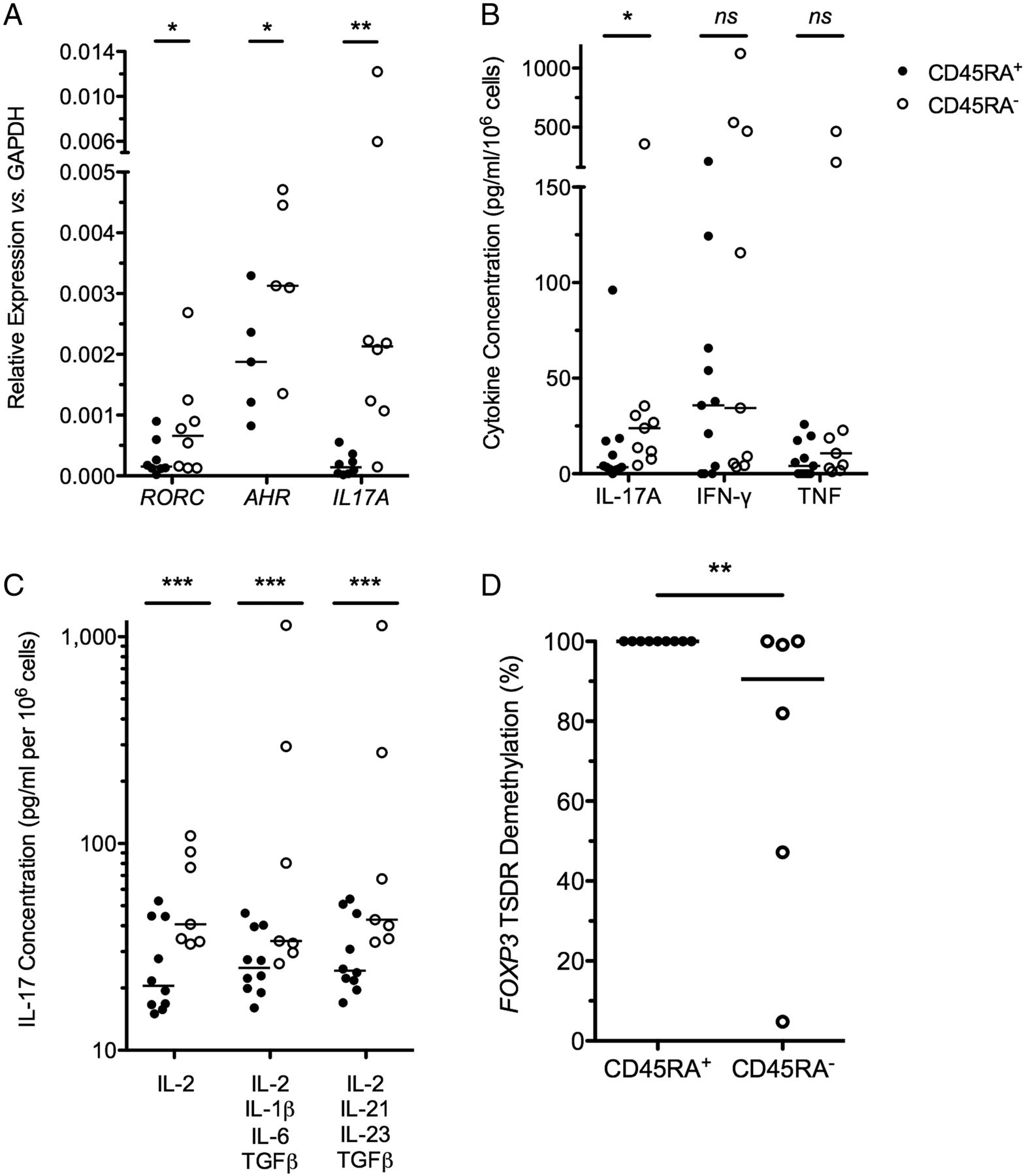
The ‘inflammatory potential’ of in vitro expanded Tregs from patients with CD was examined. Genes important in Th17 biology, including RORC, AHR and IL-17, were significantly overexpressed in CD45RA− Tregs, in comparison with expression in paired CD45RA+ Tregs (p<0.05 for each comparison, figure 2A). IL-17 secretion was also significantly different in these Treg subsets. IL-17 expression was below the limit of detection in 10/11 (91%) CD45RA+ Tregs and significantly higher in CD45RA− Tregs (p=0.02; figure 2B).

D24 CD45RA+ Tregs are resistant to IL-17 induction. (A) Relative expression of IL17A, RORC and AHR in D24 CD45RA+ and CD45RA− Tregs, relative to GAPDH; n=16, bar at median. (B) D24 Treg IL-17, IFN-γ and TNF secretion in 24 h culture supernatants; n=20, bar at median. (C) IL-17 detected by ELISA from 5-day culture supernatants of D24 Tregs cultured in the absence of rapamycin but with supplemental IL-2 alone, a cocktail of IL-2, IL-1, IL-6 and TGF-β or a cocktail of IL-2, IL-21, IL-23 and TGF-β. n=17, bar at median. (D) % FOXP3 TSDR demethylation; n=15, bar at median. *p<0.05,**p<0.01,***p<0.001. Tregs, thymus-derived regulatory T cells; IL, interleukin; IFN, interferon; TNF, tumour necrosis factor; TGF, transforming growth factor; TSDR, Treg-specific demethylated region; NS, not significant.
The potential of in vitro expanded Tregs to turn on an inflammatory programme following exposure to Th17-inducing cytokines, as occurs in vitro in Tregs isolated from blood,39–41 was examined. D24 Tregs were washed and cultured for a further 5 days in the presence of IL-2 alone, or Th17-inducing cytokines (IL-2, IL-1, IL-6 and TGF-β or IL-2, IL-21, IL-23 and TGF-β; figure 2C). These proinflammatory cytokines failed to induce IL-17 production by CD45RA+ Tregs. In contrast, IL-17 production by CD45RA− Tregs was 3-fold higher than CD45RA+ Tregs in neutral conditions (IL-2 alone) and 10-fold higher in skewing conditions (p<0.001 each comparison).
To ensure that phenotypic stability of CD45RA+ Tregs correlated with an epigenetically stable FOXP3 locus, we determined the methylation status of the FOXP3 ‘TSDR’ (figure 2D). We found the TSDR to be completely demethylated in all CD45RA+ Treg lines tested (100%; n=9), suggesting an epigenetically stable FOXP3 locus in CD45RA+ Tregs even after 24d of in vitro expansion. In contrast, variable degrees of TSDR demethylation were seen in CD45RA− Treg lines (median (IQR) of 90.6% (36.6%–100%); n=6; p=0.008).
In vitro expanded CD45RA+ Tregs express homing receptors for gut and lymphoid tissue
The ability of in vitro expanded Tregs to home to relevant immune niches, where they may suppress inflammation, is thought to be critical for cell therapy. Consequently, the expression of gut homing receptors on in vitro expanded Tregs was examined by FACS (figure 3A, B). We found that D24 CD45RA+ Tregs modestly expressed α4β7 integrin and CCR6 (20.8%±7.8% and 12.2%±7.9%, respectively) and did not express CCR9. Both CD62L (84.8%±20.6%; p=0.04 vs CD45RA−) and CCR7 (92.1%±12.8%; p=0.03) required for lymph node homing were more highly expressed in CD45RA+ Tregs than CD45RA− Tregs. CCR4 (95.4%±4.2%) was also highly expressed.

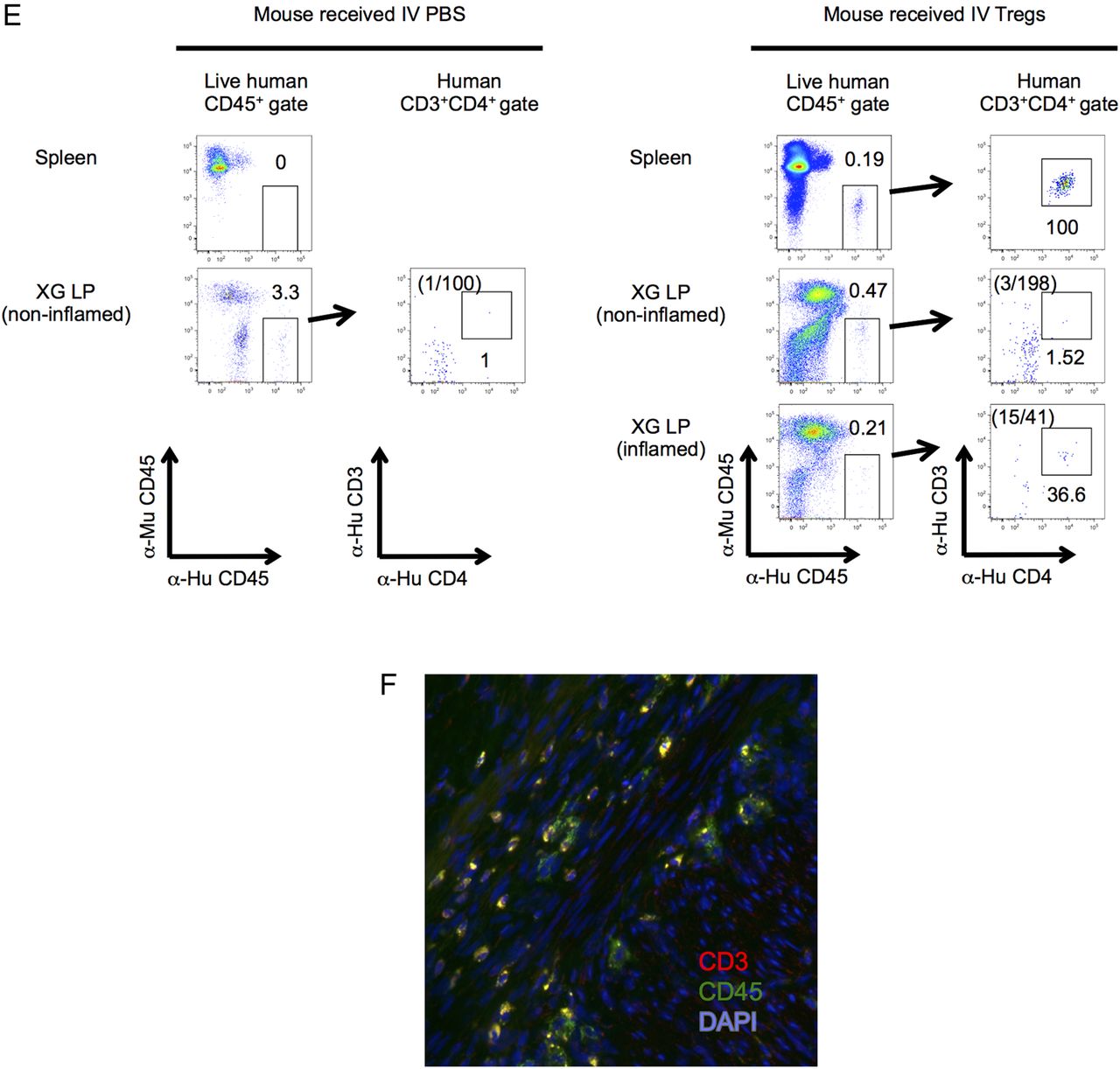
D24 CD45RA+ Tregs express gut and lymphoid homing receptors and home to inflamed human LP in a C.B-17 severe combined immunodeficiency (SCID) mouse human intestinal XG model. (A) Representative FACS plots illustrating gut and lymphoid homing receptor expression on D24 CD45RA+ Tregs (bold line). Gates were drawn on the basis of fully stained CD4+ lymphocytes (filled) and fluorochrome minus one (FMO) controls. (B) Dot plots showing expression of intestinal and lymphoid homing receptors in D24 CD45RA+ and CD45RA− Tregs. n=17; *p<0.05. (C) Design of the XG mouse experiment. (D) Left panel: mature XGs (circled) are visible subcutaneously on the dorsum of the mouse. Right panel: dorsal skin has been removed in an anaesthetised mouse to reveal the mucus-filled XG in situ (right panel). Microscopic images of the XG are shown in online supplementary figure S4A. (E) FACS plots showing live human CD45+CD3+CD4+ events in single cell suspensions prepared from murine spleen, non-inflamed and inflamed XGs, 24 h after intravenous phosphate buffered saline (PBS) (left panels) or adoptive transfer of Tregs (right panels). The absolute numbers of CD3+CD4+ events in the XG human CD45+ gates are highlighted. The gating strategy is illustrated in online supplementary figure S4B. (F) Immunofluorescence staining of XG cryosections with antihuman CD3 (red), antihuman CD45 (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue). (E and F) Representative of two independent experiments. Tregs, thymus-derived regulatory T cells; LP, lamina propria; XG, xenograft; FACS, fluorescence-activated cell sorting; EPEC, enteropathogenic Escherichia coli; NS, not significant.
Adoptively transferred CD45RA+ Tregs home to inflamed human small intestine in a C.B-17 SCID human SB xenotransplant model
In view of the favourable phenotype of CD45RA+ Tregs as a candidate cell therapy, we next sought to determine whether these cells could home to inflamed human SB in vivo. D24 CD45RA+ Tregs were administered to a C.B-17 SCID mouse bearing human small intestinal xenotransplants and homing assessed 24 h later (figure 3C, D). Intraluminal injection with enteropathogenic Escherichia coli was used to induce mucosal inflammation (see online supplementary figure S4A). Following adoptive transfer, human CD45+CD3+CD4+ cells were detected in mouse spleen and inflamed human SB LP by FACS (see figure 3E; gating strategy online supplementary figure S4B), indicating that adoptively transferred CD45RA+ Tregs homed to inflamed human SB LP in this model. This was confirmed by the detection of human CD45+CD3+ cells in inflamed human SB LP by immunofluorescence (figure 3F). We previously showed that human fetal SB contains a population of CD3−CD7+ cells that persist following xenotransplantation.53 Human CD45+CD3− events were also detected in non-inflamed human SB LP in both mice that received intravenous PBS and intravenous Tregs (figure 3E), suggesting that a population of long-lived human immune cells was co-transferred with the human SB transplant.
In vitro expansion enhances the in vitro suppressive ability of CD45RA+ Tregs
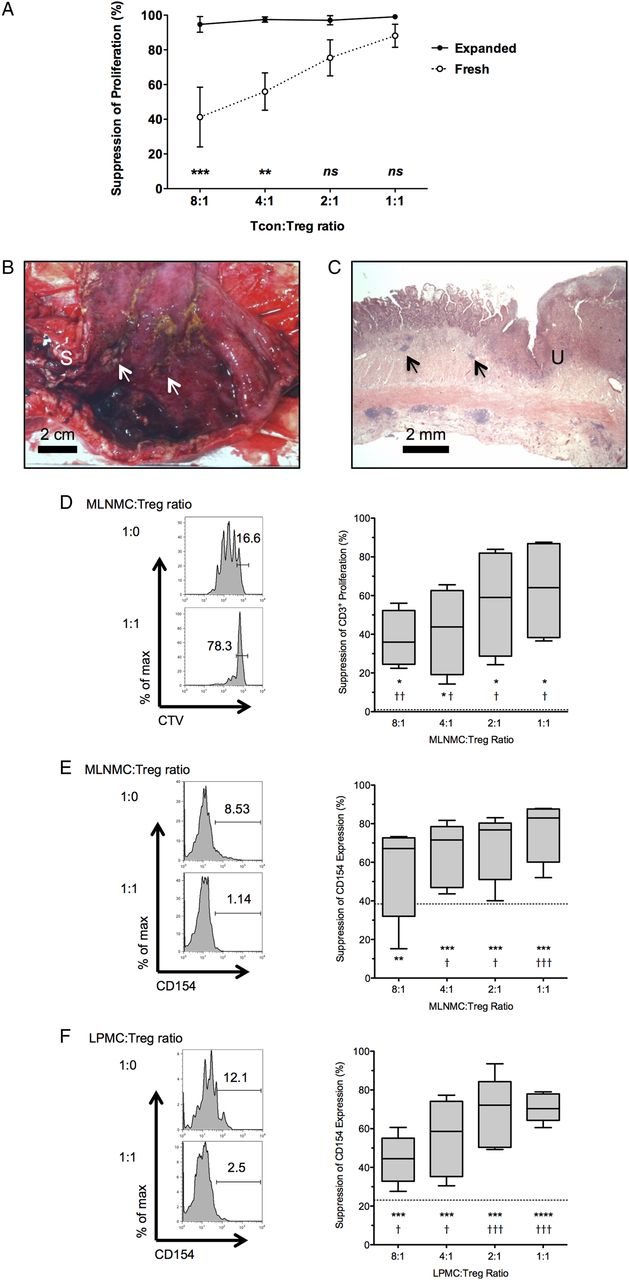
LP Tcons from inflamed CD mucosa are resistant to in vitro suppression by autologous LP Tregs.35 ,36 Consequently, it is possible that in vitro expanded Tregs will need an enhanced suppressive function in order to be successful as a future cell-based therapy. Expansion with supplemental rapamycin enhances the in vitro suppressive ability of Tregs from patients with ESRF, SLE, RA, MS and asthma.26 ,38 In order to determine if in vitro expansion enhanced Treg function in patients with CD, freshly isolated CD4+CD25hiCD127loCD45RA+ Tregs or D24 CD45RA+ Tregs that were expanded in vitro from these FACS-sorted CD4+CD25hiCD127loCD45RA+ precursors were co-cultured with allogeneic Carboxyfluorescein succinimidyl ester (CFSE)-labelled CD4+CD25− Tcons (n=3 independent experiments; cells from the same lot of single-donor, freeze-thawed Tcons for each experiment). D24 CD45RA+ Tregs suppressed Tcon proliferation to a greater degree than the freshly isolated CD4+CD25hiCD127loCD45RA+ Tregs from which they were expanded, at both a 4:1 and 8:1 Tcon:Treg ratio (p<0.01 and p<0.001, respectively; figure 4A). This suggests that in vitro expansion enhances the suppressive ability of D24 CD45RA+ Tregs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro expanded CD45RA+ Tregs suppress CD3+ T cell responses from inflamed Crohn's MLN and LP. (A) Suppression of proliferation of a single lot of freeze-thawed, allogeneic Tcons by freshly isolated PB CD4+CD25hiCD127loCD45RA+ Tregs, or D24 CD45RA+ Tregs that were expanded in vitro from these freshly isolated precursors. Pooled data from three sets of freshly isolated PB Tregs and subsequently expanded Treg populations. Data points are mean±SEM. (B) Fresh ileal resection specimen opened longitudinally to show ileal stricture (marked ‘S’) and proximal inflamed, haemorrhagic mucosa with deep ulceration (arrows). Scale bar: 2 cm. (C) Representative microscopic image from this resection showing mucosal distortion, ulceration (marked ‘U’) and transmural inflammation, including lymphoid aggregates (arrows). 12.5× H&E. Scale bar: 2 mm. (D) Representative FACS plots gated on live CD3+ events, showing proliferation of MLN Tcons cultured alone (top left panel) or with Tregs at a 1:1 MLNMC:Treg ratio (bottom left panel). Pooled data showing Treg-mediated suppression of MLN CD3+ proliferation (right panel, n=5). Box and whisker plot shows median, IQR and range. (E) Representative FACS plots gated on live MLN CD3+ events showing CD154 expression on MLN Tcons cultured alone (top left panel) or with Tregs at a 1:1 MLNMC:Treg ratio (bottom left panel). Pooled data showing Treg-mediated suppression of CD154 expression in live MLN CD3+ cells (right panel, n=5). (F) Representative FACS plots gated on live LP CD3+ events showing CD154 expression on LP Tcons cultured alone (top left panel) or with Tregs at a 1:1 LPMC:Treg ratio (bottom left panel). Pooled data showing Treg-mediated suppression of CD154 expression in live LP CD3+ cells (right panel, n=5). (D–F) Dotted line shows non-specific suppression from ‘2X control’. Comparisons between observed suppression and non-specific suppression (†p<0.05, ††p<0.01, †††p<0.001, ††††p<0.0001) and observed suppression and no suppression (zero, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001) are shown. Tregs, thymus-derived regulatory T cells; MLN, Mesenteric lymph node; LP, lamina propria; Tcons, conventional CD4+CD25lo/int T cells; PB, peripheral blood; FACS, fluorescence-activated cell sorting; MLNMC, MLN mononuclear cell; LPMC, LP mononuclear cell; CTV, Cell Trace Violet.
In vitro expanded CD45RA+ Tregs suppress proliferation and activation of MLN and LP T cells in active CD
We next wished to determine if D24 CD45RA+ Tregs could suppress activation and proliferation of Tcons taken from the MLN and LP of patients with CD (figure 4B, C). MLNMCs were co-cultured with Tregs and CD3+ proliferation assessed at 96 h. Dose-dependent Treg-mediated suppression of MLN CD3+ proliferation was seen at each MLNMC:Treg ratio (figure 4D). We were unable to demonstrate in vitro suppression of LPMC CD3+ proliferation with this technique, as both freshly isolated and freeze-thawed LPMCs obtained from inflamed CD mucosa died prior to acquisition at 96 h (n=4 independent experiments; see online supplementary figures S5 and S6).
We recently validated a novel co-culture assay for the assessment of in vitro expanded Treg function. This takes advantage of Treg-mediated suppression of the early activation marker CD154 (CD40 L) on Tcons at 7 h, which correlates with Treg-mediated suppression of CFSE dilution and cytokine expression in Tcons at 96h25 ,49 Significant dose-dependent suppression of CD154 expression in MLN and LP T cells was observed (figure 4E, F), demonstrating that in vitro expanded D24 CD45RA+ Tregs suppress early activation of MLN and LP Tcons in vitro.
Discussion
There remains an unmet need to develop novel therapies for CD, as current drug treatments frequently fail to maintain long-term remission and may be complicated by significant side effects. Cellular therapies are emerging as potentially attractive therapeutic strategies. Tregs are effective in preclinical models of colitis2 ,6 and phase 1 clinical trials suggest that in vitro expanded Tregs are safe in the prophylaxis and treatment of GvHD12 ,24 and type 1 diabetes.18 We built on recent work to describe a method for isolation and expansion of Tregs from Crohn's blood that is readily transferable to a GMP background and addresses several barriers to the use of expanded Tregs as an autologous cell-based therapy in this important disease.
Tregs can be selected and expanded in vitro to clinically useful numbers under both R&D-grade,11 13 ,16 ,21 ,23 ,26 and GMP conditions12 ,18 ,24 retaining an in vitro suppressive function before infusion into humans. We showed that it is feasible to do the same using Tregs obtained from Crohn's blood, including patients receiving thiopurines or anti-tumor necrosis factor (TNF) medications. Even after prolonged culture, these Tregs maintained FOXP3 expression and suppressed activation of autologous T cells.
T cell lineage plasticity is well described. A major potential barrier to Treg therapy is the possibility that these cells might adopt an inflammatory phenotype and worsen inflammation on adoptive transfer. Freshly isolated thymus-derived Tregs from both mice and humans can express proinflammatory cytokines and transcription factors (TF) canonical to effector CD4+ lineages, including IL-1739–41 and IFN-γ,44 both of which are implicated in CD pathogenesis. Indeed, IL-17+FOXP3+ Tregs have been identified in non-inflamed human blood and lymphoid tissue,40 and inflamed Crohn's mucosa.43 While there is some evidence that plastic cytokine and TF expression may license efficient Treg homing to, and suppression of, Th1-mediated and Th17-mediated inflammation,44 ,56 this may also lead to the generation of Tregs with an effector phenotype that contribute to inflammation.
We and others have demonstrated that in vitro expanded Tregs cultured in the presence of rapamycin have enhanced phenotypic stability.13 ,21 We show that as well as retaining their suppressive capacity, CD45RA+ rTregs expanded from the blood of patients with CD in the presence of rapamycin do not express IL-17A or other Th17-related genes, even following exposure to proinflammatory cytokines that they would likely meet in inflamed intestinal mucosa. These data corroborate data from Hoffmann et al17 ,47 in healthy controls, showing that expanded CD45RA+ Tregs are resistant to the induction of proinflammatory cytokines on stimulation and highly express CD62L and CCR7, which are associated with phenotypic stability.
Freshly isolated CD45RA+ rTregs have an epigenetically stable FOXP3 locus with extensive TSDR demethylation.46 TSDR demethylation correlates with stable FOXP3 expression in vitro50 and Treg-mediated protection from autoimmunity in vivo57 in humans. However, the significance of TSDR demethylation for in vitro expanded Tregs is poorly understood. Barzaghi et al57 recently described a cohort of patients with ‘Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked syndrome (IPEX)-like syndrome’, severe multisystem autoimmunity in the absence of identifiable mutations in molecules implicated in Treg function, with decreased TSDR demethylation despite normal Treg numbers and in vitro suppression. This suggests that ex vivo expanded CD45RA− Tregs, with incomplete TSDR demethylation, may have suboptimal biological activity in vivo, despite suppressive function in vitro. These data also suggest that CD45RA+ Tregs are more likely to retain phenotypic stability and are less likely to acquire an effector phenotype than CD45RA− Tregs, consistent with a more favourable safety profile of this Treg subset as a cell-based therapy for CD.
In order to be therapeutically effective, adoptively transferred Tregs may need to traffic to intestinal lymphoid tissue or LP. Some groups have taken advantage of TCRs specific for luminal antigens to direct Tregs to the intestinal mucosa, such as IL-10-producing T cell clones with ovalbumin-specific TCRs,58 or T cells with transgenic Cbir1 flagellin-specific TCRs.59 Alternatively, Treg expansion in the presence of ATRA induces α4β7 integrin expression but also increases effector cytokine expression, such as IL-17 and IFN-γ, potentially limiting its use in GMP cell expansion.13 ,21 We show that CD45RA+ Tregs expanded in the presence of IL-2 and rapamycin highly express CD62L and CCR7, allowing homing to, and anatomical orientation within lymphoid tissue.60 ,61 Treg CD62L expression is also required for Treg-mediated cure of GvHD.30 CD45RA+ Tregs also expressed CCR4, required for Treg-mediated prevention of CD45RBhi colitis.62 Interestingly, murine Tregs do not need to home to intestinal LP to prevent CD45RBhi adoptive transfer colitis. β7 integrin-null Tregs home to MLN and prevent colitis in this model, despite almost undetectable LP homing.63 Consequently, the ability to home to MLN is highly desirable in potentially therapeutic cells.
CD45RA+ Tregs also express α4β7 integrin and CXC motif receptor 3 (CXCR3), indicating an ability to home to LP and sites of inflammation, respectively. Moreover, we used a human small intestinal xenotransplant model to show, for the first time, that in vitro expanded CD45RA+ Tregs from patients with CD home to inflamed human gut in vivo. Xenotransplanted SB segments develop into tissue that is morphologically and functionally identical to normal gut and is capable of peristalsis and nutrient absorption.53 ,54 The xenografts also possess a chimeric endothelium that expresses human MadCAM-1.64 This is the first demonstration that this model can be used in the assessment of immune cell homing.
Xenograft-bearing mice received rhIL-2 in order to support survival of adoptively transferred human Tregs,23 as murine IL-2 is less efficient at promoting proliferation of human T cells than rhIL-2, despite cross-reactivity.65 As recent phase 1 trials of in vitro expanded Tregs in GvHD and type 1 diabetes mellitus showed signs of clinical efficacy without supplemental rhIL-2, it is likely that this is a feature of the experimental system and will not be required in clinical trials in Inflammatory bowel disease (IBD).12 ,18 ,24
Future work will include ‘humanising’ xenograft-bearing mice and developing additional techniques to induce xenograft inflammation, thus allowing us to assess the functional impact of CD45RA+ Tregs on gut inflammation. The percentage of LP human T cells that could be recovered from human bowel transplants was relatively modest compared with the percentage of T cells recovered from the spleen. Given that the expression of the gut homing integrin α4β7 was only expressed on ∼20% of the purified Tregs, future work may need to address methods to increase α4β7 expression, such as the use of retinoic acid, as we have previously shown.21
An additional barrier to Treg therapy in CD is that effector T cells from the diseased mucosa of patients with CD may be resistant to the suppressive action of Tregs. Indeed, we previously showed that Tcons isolated from inflamed Crohn's mucosa are relatively resistant to Treg-mediated suppression, due to overexpression of Smad7, an inhibitor of TGF-β signalling.35 ,36 In this study, we utilised Tregs cultured in the presence of rapamycin, which has been shown to enhance the suppressive ability of in vitro expanded Tregs, compared with Tregs freshly isolated from the same donor26 ,38 and show that in vitro expansion enhances the suppressive ability of Tregs obtained from CD PB. Rapamycin-expanded CD45RA+ Tregs effectively suppress both MLN and LP T cells obtained from inflamed Crohn's resection specimens. These data suggest that in vitro expanded CD45RA+ Tregs may modulate immune responses in niches directly relevant to the pathogenesis of CD. Tregs use multiple mechanisms to suppress in vitro and in vivo, including contact-dependent mechanisms (CTLA-4, perforin-granzyme B) and contact-independent mechanisms (IL-10, TGF-β, extracellular ATPase activity via CD39/CD73,etc). Sakaguchi et al10 has proposed a multistep model of in vitro suppression that initially requires cell-cell contact but is subsequently contact independent. The mechanism of suppression of erstwhile ‘resistant’ mucosal Tcons by in vitro expanded Tregs is currently unknown and will be the subject of further study. In addition, not all of the patients in this study had active disease, so it will be important to extend these data further to broaden the therapeutic relevance of these findings. However, a substantial proportion of the patients in this study did have evidence of disease activity (n=5/13), which did not affect either Treg expansion or function.
In conclusion, we have shown that in vitro expanded CD45RA+ Tregs are likely to be the most suitable Treg subset for cellular therapeutics in CD. This subset is readily expandable to sufficiently high numbers under conditions that are readily transferable to GMP, for clinical use. They express an appropriate repertoire of homing receptors for MLN and gut, and effectively traffic to inflamed gut in vivo. As well as retaining powerful suppressive properties, these cells show little or no capacity for plasticity towards a potentially harmful effector phenotype, which correlates with an epigenetically stable FOXP3 locus. This study addresses many of the perceived barriers to Treg cell treatment for CD and paves the way for a clinical trial of in vitro expanded CD45RA+ Tregs in this therapeutically challenging disease.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
JBC and CS contributed equally.
Correction notice This article has been corrected since it published online first. The results section has been updated.
Contributors JBC designed and executed the experiments, analysed and interpreted the data and wrote the manuscript. MJE, IS, AV, CS, RG, EM and JWL designed and executed experiments and contributed to the manuscript. ES, NP, HF and JKH provided intellectual input and contributed to the manuscript. JBC, GML, PMI, JDS and SY developed the research infrastructure and governance for human sample collection. PMI, JDS and SY provided ongoing clinical care and intellectual input, and contributed to the manuscript. TTM, MPH-F, NYS and GL designed the experiments, interpreted data and wrote the manuscript. GML is the senior author and guarantor of this manuscript.
Funding This study was supported by grants awarded by the National Institute for Health Research (JBC, GML; grant number DRF/2009/02/22), Guy's and St Thomas’ Charity (JBC, MPH-F, JDS, GML; grant number R090707), the Academy of Medical Sciences (JBC; Daniel Turnberg Memorial Fund) the Medical Research Council, UK (GML, TTM; grant number G0802068; GML, JKH, grant number MR/K002996/1; MPH-F, grant numbers G0801537/ID: 88245 and MR/J006742/1), the Wellcome Trust (NP, GML, TTM, grant number WT088747MA, GML, grant number 091009), the European Union 7th Framework Programme (EU FP7) (GLom, CS, MPH-F; The ONE study; reference 260687; MPH-F; BIO-DRIM; reference 305147) and King's Health Partners Research and Development Challenge Fund, Guy's and St Thomas’ Charity (CS; grant number R1405170). Research was also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ National Health Service (NHS) Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Competing interests None.
Ethics approval South East London REC 2 (10/H0804/65). East London REC 2 (10/H0704/74). Ethics Committee for Animal Experimentation, Hebrew University of Jerusalem (MD-11-12692-4). Helsinki Committee of the Hadassah University Hospital (81-23/04/04).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All available data are included in this manuscript.