Article Text

Abstract
Aims The activation of β-catenin signalling is a key step in intestinal tumorigenesis. Interplay between the β-catenin and Notch pathways during tumorigenesis has been reported, but the mechanisms involved and the role of Notch remain unclear.
Methods Notch status was analysed by studying expression of the Notch effector Hes1 and Notch ligands/receptors in human colorectal cancer (CRC) and mouse models of Apc mutation. A genetic approach was used, deleting the Apc and RBP-J or Atoh1 genes in murine intestine. CRC cell lines were used to analyse the control of Hes1 and Atoh1 by β-catenin signalling.
Results Notch signalling was found to be activated downstream from β-catenin. It was rapidly induced and maintained throughout tumorigenesis. Hes1 induction was mediated by β-catenin and resulted from both the induction of the Notch ligand/receptor and Notch-independent control of the Hes1 promoter by β-catenin. Surprisingly, the strong phenotype of unrestricted proliferation and impaired differentiation induced by acute Apc deletion in the intestine was not rescued by conditional Notch inactivation. Hyperactivation of β-catenin signalling overrode the forced differention induced by Notch inhibition, through the downregulation of Atoh1, a key secretory determinant factor downstream of Notch. This process involves glycogen synthase kinase 3 β (GSK3β) and proteasome-mediated degradation. The restoration of Atoh1 expression in CRC cell lines displaying β-catenin activation was sufficient to increase goblet cell differentiation, whereas genetic ablation of Atoh1 greatly increased tumour formation in Apc mutant mice.
Conclusion Notch signalling is a downstream target of β-catenin hyperactivation in intestinal tumorigenesis. However, its inhibition had no tumour suppressor effect in the context of acute β-catenin activation probably due to the downregulation of Atoh1. This finding calls into question the use of γ-secretase inhibitors for the treatment of CRC and suggests that the restoration of Atoh1 expression in CRC should be considered as a therapeutic approach.
- Intestine
- β-catenin
- RBP-J
- Hes1
- Atoh1
- proliferation
- differentiation
- cell proliferation
- cell signalling
- colorectal cancer genes
- differentiation
- molecular carcinogenesis
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Intestine
- β-catenin
- RBP-J
- Hes1
- Atoh1
- proliferation
- differentiation
- cell proliferation
- cell signalling
- colorectal cancer genes
- differentiation
- molecular carcinogenesis
Significance of this study
What is already known about this subject?
Inhibition of the Notch signalling pathway using γ-secretase inhibitors can force differentiation of intestinal tumours induced by Apc loss into goblet cells. Thereby, γ-secretase inhibitors have been proposed as a rationale for treatment of intestinal cancers.
Accordingly, interplay between the β-catenin and Notch pathways during tumorigenesis has been reported, but the mechanisms involved and the role of Notch remain unclear.
What are the new findings?
By analysing murine and human samples of all steps of intestinal tumorigenesis, we found that Notch signalling activation is an immediate effect of Wnt/β-catenin deregulation and is maintained throughout all the intestinal tumorigenesis.
In addition, we showed that, in intestinal tumorigenesis, the induction of Hes1 a well-known Notch target gene, was mediated directly by β-catenin signalling activation. Thus, Hes1 expression results from both a Notch-dependent and a Notch-independent signalling in intestinal tumorigenesis.
Surprisingly, the strong phenotype of unrestricted proliferation and impaired differentiation induced by acute Apc deletion in the intestine was not rescued by conditional Notch inactivation. Hyperactivation of β-catenin signalling overrode the forced differentiation induced by Notch inhibition, through the downregulation of Atoh1, a key secretory determinant factor downstream of Notch. This downregulation is critical to the oncogenic outcome mediated by β-catenin because it prevents the differentiation of cancer cells.
How might it impact on clinical practice in the foreseeable future?
Our findings have important clinical implications, as they call into question the utility of γ-secretase inhibitors as a treatment for colorectal cancer and suggest that the stabilisation of Atoh1 may be of therapeutic importance.
Introduction
The Apc gene encodes a tumour suppressor linked to 80% of cases of sporadic colorectal cancer (CRC) and involved in an inherited syndrome, familial adenomatous polyposis syndrome.1 We and others have clearly shown that the inactivation of Apc is sufficient to initiate intestinal tumorigenesis.2 3 One of the major effects of Apc loss is the aberrant activation of Wnt/β-catenin signalling. The stabilised β-catenin is translocated to the nucleus, where it forms a complex with Tcf/Lef transcription factors, activating a genetic programme leading to an imbalance between cell proliferation and differentiation that triggers tumorigenesis.1
The Notch pathway is also known to be involved in the control of cell fate choice between proliferation and differentiation in intestinal epithelial lineages, and thus plays a major role in intestinal homeostasis and tumorigenesis.1 The genes of the Notch family encode transmembrane receptors. Interactions between Notch receptors and Delta or Jagged ligands result in a conformational change, followed by two proteolytic steps. These cleavages release the Notch intracellular domain, which forms a complex with the transcription factor RBP-J to activate the transcription of downstream targets, such as the Hes1 gene.4 Loss-of-function studies have shown that Notch signalling upregulates absorptive cell differentiation and downregulates the differentiation of secretory lineages, including goblet cells, Paneth cells and enteroendocrine cells.5 6 Cell fate seems to be directly controlled by two main Notch target genes: Hes1, a positive target, which stimulates the differentiation of progenitors into absorptive enterocytes; and Atoh1, a negative target gene that commits the progenitor to a secretory cell fate. These studies, together with gain-of-function studies, have also provided evidence that Notch signalling is required for the maintenance of crypt progenitor cells in an undifferentiated and proliferative state, suggesting a possible role in tumorigenesis.7–9 However, whereas Wnt/β-catenin signalling is well known to play multiple roles in intestinal tumorigenesis, the role of Notch in this process remains unclear.
We investigated the interplay between the Wnt/β-catenin and Notch signalling pathways in intestinal tumorigenesis. We first evaluated Notch signalling status during intestinal tumorigenesis in humans and mice. We used several mouse models of Apc mutation reproducing all steps of intestinal tumorigenesis, from initiation to the more advanced stages, with the aim of determining when the Notch signalling pathway was activated during this process. For studies of the impact of Notch signalling during the intestinal tumorigenesis induced by Apc loss, we simultaneously deleted both the Apc and RBP-J or Atoh1 genes in the intestinal epithelium.
We found that Notch acted downstream from Wnt/β-catenin at all stages of intestinal tumorigenesis and, surprisingly, that Notch/RBP-J signalling was dispensable for the initiation of β-catenin-mediated intestinal tumorigenesis. Our results indicate that the inability of Notch/RBP-J inhibition to direct differentiation was directly linked to post-transcriptional control of Atoh1 by active β-catenin signalling. Finally, we found that ablation of the Atoh1 gene strongly enhanced tumour formation in Apc mutant mice. Thus, aberrant β-catenin signalling has two main consequences in intestinal epithelial cells: stimulation of the proliferation of stem cells and progenitors, and inhibition of their differentiation.
Materials and methods
Collection of samples from patients
Colorectal adenocarcinomas and corresponding normal tissue samples were collected at the Hotel-Dieu and Cochin Hospitals (France). The tissue samples were obtained with informed, written consent from the patients and the approval of the local ethics committee. A tissue microarray containing 55 colon adenocarcinomas was analysed with a microarrayer (Beecher Instruments, Wisconsin, USA).
Animal generation
All experiments involving mice were carried out in accordance with French government regulations. Mice were housed in conventional conditions. The generation of mutant mice and treatments are described in the supplementary material.
Histology, immunohistochemistry and in situ hybridisation
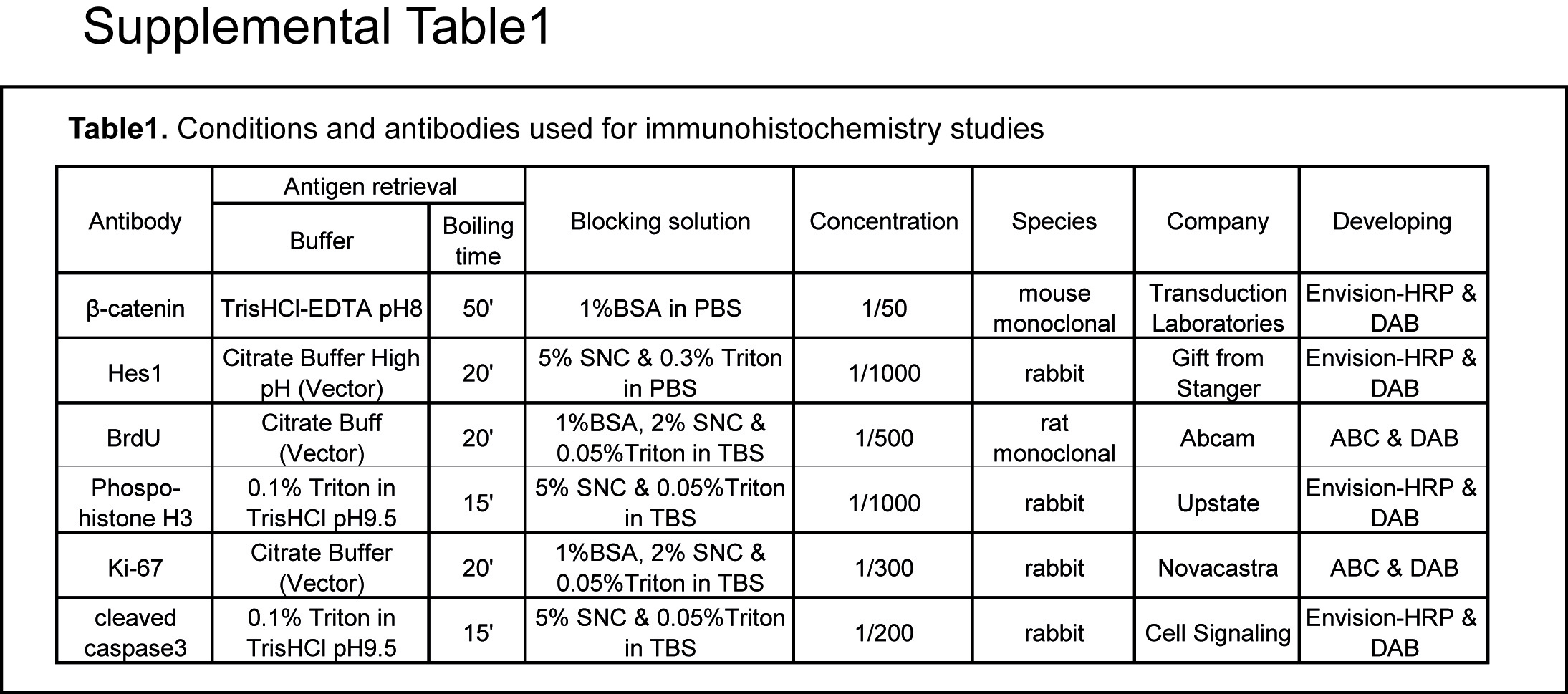
For histological, immunohistochemical and in situ hybridisation methods, see the supplementary materials and Supplementary table 1.
Molecular and biochemical analyses
Detailed descriptions of procedures are provided in the supplementary material.
In silico promoter analysis
For the identification of phylogenetically conserved Tcf-binding sites, the Hes1 5′-flanking regions were analysed with Gene2Promoter, DiAlign TF and MatInspector software from the Genomatix database.
Statistical analysis
The significance of the differences observed was assessed with the non-parametric Wilcoxon matched-pairs test for human samples and two-tailed Student t tests for mouse samples and in vitro experiments. Detailed descriptions are provided in the supplementary materials.
Results
Hes1 expression during human and murine intestinal tumorigenesis
We assessed the potential role of Notch signalling in human CRC by first investigating the expression of Hes1, a well-known Notch target gene, in 14 random CRC cases and in patient-matched normal tissues, by quantitative reverse transcription–PCR (RT–PCR). Most of the adenocarcinomas had significantly higher levels of Hes1 mRNA than the matched colon samples (figure 1A). Consistent with this finding, immunohistochemical studies of human adenocarcinomas showed strong nuclear staining for both β-catenin and Hes1 (figure 1B). We extended our analysis of Hes1 expression in murine adenomas developed spontaneously in Apc+/− mice.10 We also observed significantly higher levels of Hes1 mRNA and strong nuclear expression of Hes1 in murine adenomas (figure 1C,D). These observations suggest that Notch signalling is activated throughout intestinal tumorigenesis, in both humans and mice.

Induction of Hes1 expression in human and murine intestinal carcinogenesis. Analysis of Hes1 mRNA levels by quantitative reverse transcription–PCR (RT–PCR) in human colorectal cancer (CRC) and in normal tissue (A). Immunostaining for Hes1 and β-catenin in human CRC and in normal tissue (B). Scale bars, 200 μm. Analysis of Hes1 expression by quantitative RT–PCR (C) and immunohistochemistry (D) in normal tissue and adenomas from APC+/− mice.
Mutational analyses of the Notch genes in human CRC
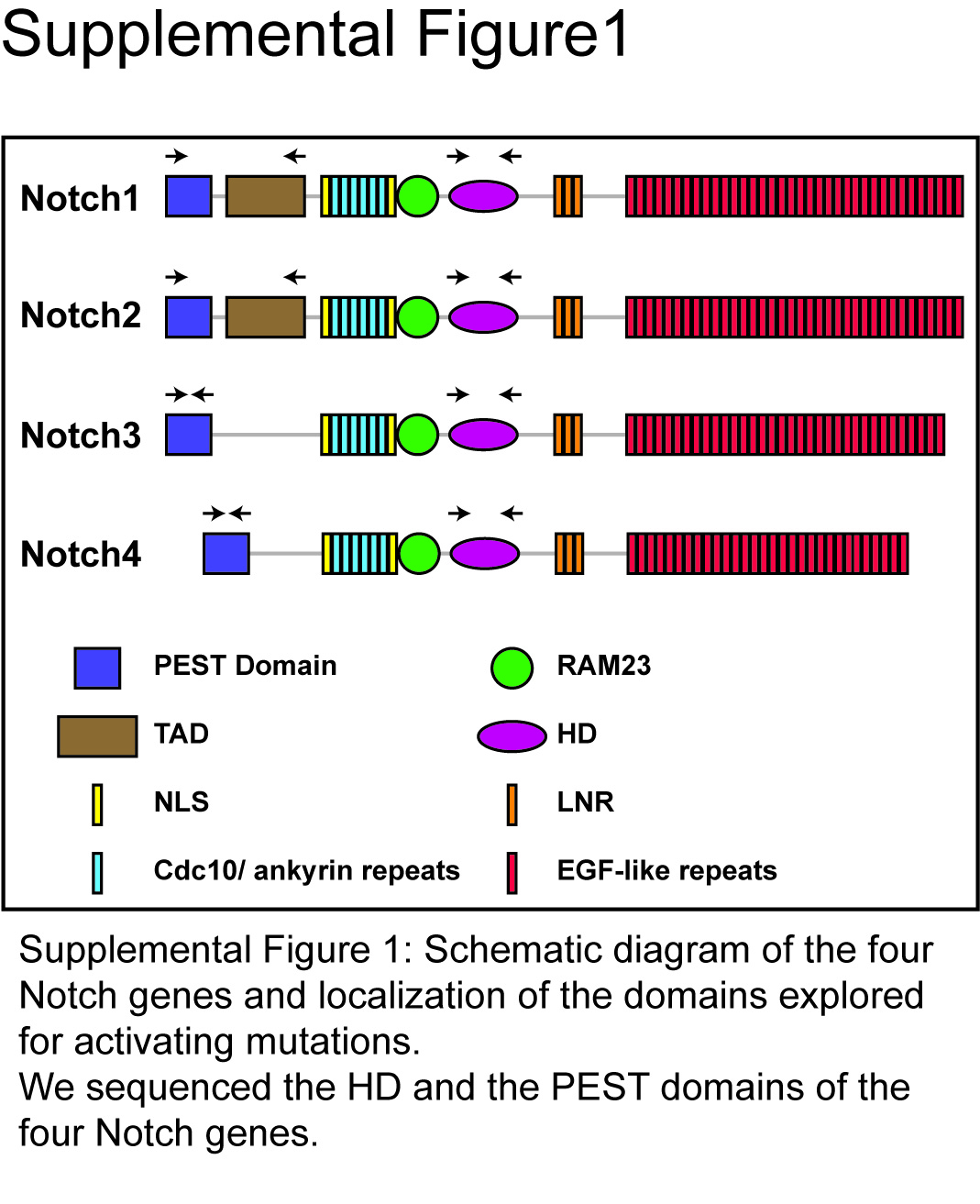
One possible mechanism of Hes1 induction during oncogenesis is the occurrence of activating mutations in Notch receptors. Such mutations were first described in the Notch1 gene in T cell acute lymphoblastic leukaemia.11 They affect the intracellular heterodimerisation domain and/or the C-terminal PEST domain of Notch1. We carried out a sequencing analysis to search for such somatic mutations in the Notch1, 2, 3 and 4 genes in 19 CRCs and patient-matched normal tissues (Supplementary figure 1 online). We found no such point mutations in any of the CRCs analysed (data not shown).
Loss of Apc results in the induction of Notch signalling components
We investigated the mechanism underlying abnormal Hes1 expression in CRC further by studying expression of the various ligands and receptors involved in Notch signalling. We observed no significant expression of Dll1, Dll3 and Notch3 in normal tissue or tumours (data not shown). However, quantitative RT–PCR showed that levels of Dll4, Jag1, Jag2, Notch1 and Notch2 mRNA were higher in human adenocarcinomas than in normal tissue (figure 2A). Thus, three of the five ligands tested and two of the four receptors tested were induced in human CRC. We then carried out in situ hybridisation for Notch2 and Dll4 on a cohort of 55 CRC samples on a tissue microarray. We observed weak expression of Notch2 and Dll4 in normal colon and the overexpression of both probes in many adenocarcinomas. Notch2 was moderately and strongly induced in 49% and 29% of CRCs, respectively, and Dll4 was slightly and actively induced in 30% and 47%, respectively (figure 2B). However, we found no link between the expression levels of the Notch2 and Dll4 genes in tumours, suggesting that stochastic events may determine the level of expression of these genes. Quantitative RT–PCR also showed an upregulation of Notch ligands and receptors in murine adenomas from Apc+/− mice (figure 2C).

Induction of Notch receptors and ligands in human and murine intestinal carcinogenesis. Fold change in mRNA levels for the Notch receptors and ligands, as measured by quantitative reverse transcription–PCR (RT–PCR), in comparisons of human colorectal carcinomas with normal tissue (A). In situ hybridisation of Notch2 and Delta4 in human normal colon and in colorectal cancer (B). Quantitative RT–PCR analyses of mRNA levels for the Notch receptors and ligands in normal tissues and adenomas from APC+/− mice (C).
We then investigated whether the deregulation of Notch signalling occurred early in β-catenin-induced intestinal tumorigenesis, by analysing Hes1 expression in a conditional mouse model with acute APC loss in the adult intestinal epithelium (Apclox/loxVil-CreERT2).3 In this system, Apc was deleted in response to tamoxifen injection (Apc-null mice, Apc−/−). This system is highly efficient, with almost 100% recombination of the target allele in the small intestine of adult mice and a rapid loss of Apc function.3 This genetic deletion system did not mimic the stochastic process of tumour initiation occurring in Apc+/− mice, but it did reproduce all the phenotypes previously identified as associated with early intestinal lesions. As previously reported, 5 days after tamoxifen injection, considerable enlargement of the crypt compartment, with intense cell proliferation, apoptosis and severe defects in differentiation, was observed in the intestine of mutant mice.3 This enlargement was accompanied by Hes1 induction, at both the mRNA and protein levels (figure 3A–C). Accordingly, we observed nuclear staining for Hes1 in all dysplastic cells with nuclear β-catenin, consistent with Apc loss (figure 3C). This increase in Hes1 expression could result from hyperactive β-catenin signalling or it could occur because of an expansion of cells that normally express Hes1. To distinguish between these two hypotheses, we analysed Hes1 expression in a human CRC cell line in which hyperactive β-catenin signalling was blocked. SW480 cells, which produce a truncated form of APC that cannot target β-catenin for degradation, were transfected with two different small interfering RNAs (siRNAs) targeting β-catenin. We found that Hes1 expression was markedly decreased by β-catenin depletion (figure 3D).

Hes1 expression is correlated with Wnt/β-catenin signalling. Quantitative reverse transcription–PCR (RT–PCR) analysis of Hes1 mRNA levels in control and APC−/− mice 5 days after tamoxifen injection (A). Immunoblot analyses of Hes1 levels in three different samples from control and APC−/− mice (day 5) (B), and Hes1 and β-catenin levels in SW480 cells after transfection with two small interfering RNAs (siRNAs) against β-catenin or scramble siRNA (D). β-Actin levels were used for normalisation. Immunohistochemistry studies of Hes1 and β-catenin in control and APC−/− mice (day five) (C). Scale bars, 50 μm.
Thus, the expression of Hes1, a Notch downstream effector, increases strongly and rapidly in response to the β-catenin activation induced by Apc loss in the intestine. Notch signalling pathway activation is therefore an early, downstream event in Wnt/β-catenin-induced intestinal tumorigenesis.
β-Catenin activation promotes both direct and indirect regulation of Notch signalling
We then investigated the mechanism by which Wnt/β-catenin signalling might control Hes1 expression. Two possible mechanisms were considered: an indirect mechanism involving the activation of Notch signalling partners and a direct mechanism in which β-catenin/Tcf controls the activity of the Hes1 promoter.
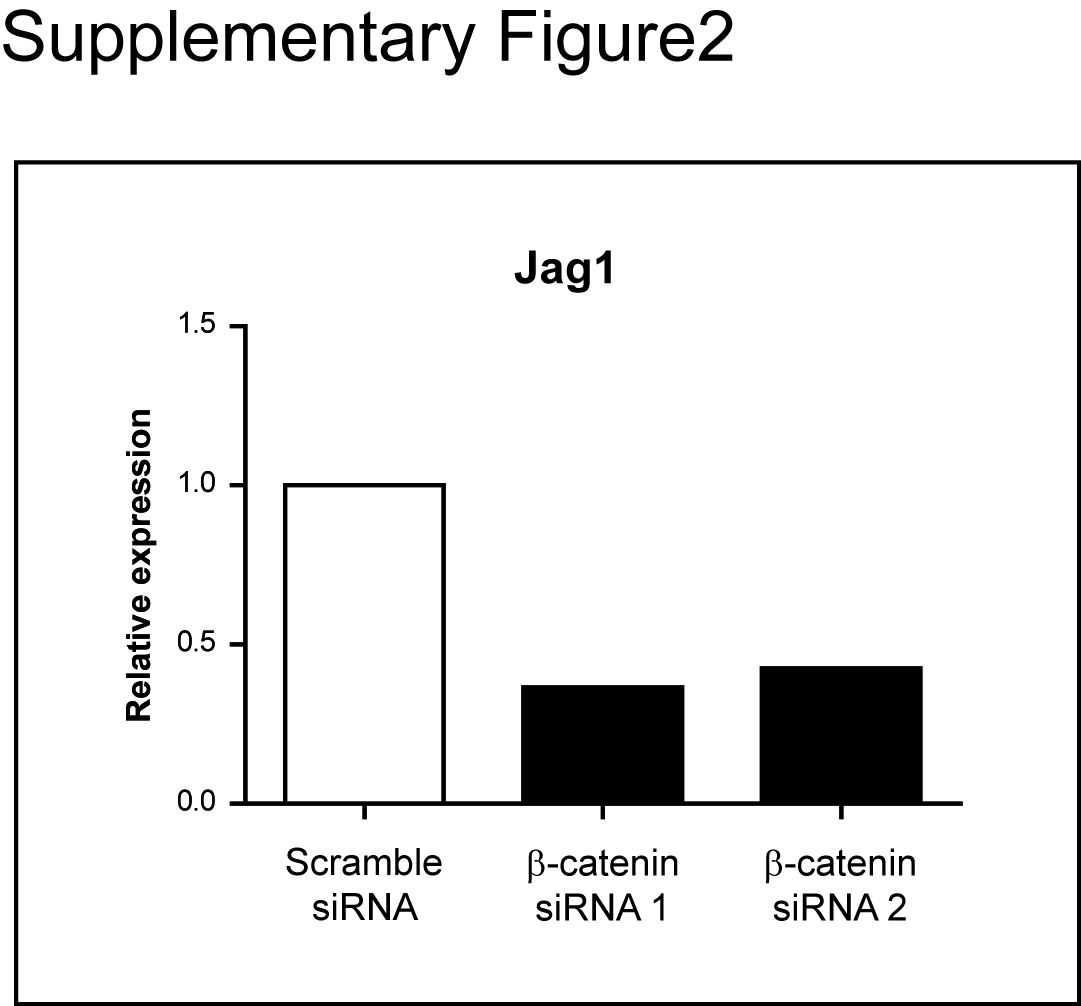
Consistent with the indirect mechanism, we observed a strong increase in the expression of Dll4, Jag1 and Jag2, and the Notch1, Notch2 and Notch4 receptors 5 days after Apc loss (figure 4A). We determined the mRNA levels of these genes before and after siRNA-mediated β-catenin downregulation in SW480 cells. Consistent with previous reports, we found that β-catenin depletion significantly inhibited only Jag1 expression (Supplementary figure 2).12 This suggests that except for Jag1 induction, the increase expression of the others ligands and the Notch receptors observed rapidly after Apc loss is not direct. An expansion of the crypt progenitor zone induced by Apc loss as an alternative explanation for the apparent increase in expression of Notch ligands and receptors could not be excluded. However, the fact that some of the ligands/receptors were strongly induced in human CRC by in situ hybridisation does not support this hypothesis.

The Hes1 promoter is regulated by Wnt/β-catenin signalling through conserved Tcf motifs. Quantitative reverse transcription–PCR (RT–PCR) analysis of mRNA levels for the Notch receptors and ligands in control and APC−/− mice (day 5) (A). Conserved Tcf-binding element motifs identified by Genomatix software in the proximal region of the Hes1 promoter of several species. The putative Tcf- and RBP-J-binding sites are represented by blue and purple boxes, respectively. Maps of the constructs used to study the transcriptional activity of the Hes1 promoter in SW480 cells. Different fragments of regulatory sequences flanking the human Hes1 promoter (Hes-2.5, Hes-0.4 and Hes-mutRBP constructs) were fused with the luciferase gene. The Hes-mutRBP construct contains only the Tcf sites, the RBP-J-binding sites having been mutated (B). All the constructs were used to transfect SW480 cells together with the Notch1 intracellular domain (NICD), the dominant-negative form of Tcf4 (ΔNTCF4) or empty vector (control) (C–E).
In addition, Genomatix software identified two conserved Tcf-binding sites in the Hes1 promoter, close to the canonical RBP-J-binding site. The conservation of these sites across species supports the hypothesis that they are functionally relevant (figure 4B). We performed luciferase reporter assays in SW480 cells to evaluate the activity of the human Hes1 promoter (Hes-2.5 and Hes-0.4 constructs) containing both the Tcf- and proximal RBP-J-binding sites. We also used the Hes-0.4-RBP-J (Hes-mutRBP) construct, which contains only the Tcf sites, the RBP-J-binding sites being mutated (figure 4B). As expected, the activation of Notch signalling by the human Notch intracellular domain induced a strong increase only in Hes-2.5 and Hes-0.4 promoter activities (figure 4C). However, similar basal levels of promoter activity were observed for the Hes-0.4 and Hes-mutRBP promoters, this activity being independent of the activation of Notch signalling (figure 4D). As these constructs contained two conserved Tcf-binding sites, the basal promoter activity observed may be linked to the aberrant Wnt/β-catenin signalling pathway. We tested this hypothesis, by analysing the effect of a dominant-negative form of Tcf4, ΔNTCF4. ΔNTCF4 induced a decrease in basal promoter activity for all constructs, whether or not they contained a functional RBP-J-binding site (figure 4E). These findings are consistent with direct control of the Hes1 promoter by Wnt/β-catenin signalling through the conserved Tcf motifs, independently of Notch activation. They are also supported by a recent genome-wide ChIP-on-chip analysis, which identified the Hes1 promoter region as a target of Tcf4.13
Thus, the induction of the downstream Notch target Hes1 in Wnt/β-catenin-mediated intestinal tumorigenesis results from two complementary processes. One is Notch dependent and involves deregulated expression of Notch ligands and receptors. The other is Notch independent and involves direct control of the Hes1 promoter by Wnt/β-catenin signalling, via the conserved Lef/Tcf sites.
RBP-J deficiency does not alter the immediate phenotype of Apc deficiency in vivo
Wnt/β-catenin and Notch cooperate during intestinal tumorigenesis,7 9 12 and our results indicate that Notch acts downstream from the Wnt cascade. We assessed the contribution of Notch signalling to the phenotype associated with Apc loss in the intestine (Apc-null mice, Apc−/−), by simultaneously deleting the RBP-J and Apc genes in the intestinal epithelium (double-mutant mice, Apc−/−RBP−/−).3 14 We chose to study the RBP-J gene, to avoid possible redundancy in the functions of Notch receptors. Based on previous published results obtained with γ-secretase inhibitors,7 we assumed that the inhibition of Notch signalling would, at least partly, rescue the defects induced by Apc loss: the increase in cell proliferation and the decrease in goblet cell differentiation.
Surprisingly, on day 5 after the first injection, we found no difference in survival between the double-mutant (Apc−/−RBP−/−) and the Apc-null mice. Both the double-mutant and the Apc-null mice became ill and 100% of the mice died within 7–8 days of the tamoxifen injection (figure 5A). No major histological difference was observed between the small intestines of the single- and double-mutant mice (figure 5B).

Loss of RBP-J does not alter the cell differentiation defects of Apc-deficient cells. Kaplan–Meier survival analysis of control (purple), APC−/− (green) and APC−/−RBP−/− mice (red) (injections are indicated by an arrow) (A). Representative H&E-stained sections of control, APC−/− and APC−/−RBP−/− mice 5 days after tamoxifen (Tam) injection (day 5) (B). PCR of DNA extracted from intestines of control and APC−/−RBP−/− mice showing the excision of the floxed RBP-J alleles (day 5) (C). Quantitative reverse transcription–PCR (RT–PCR) analysis of Atoh1, p27kip1 and p57kip2 mRNA levels in control, APC−/− and APC−/−RBP−/− mice (day 5) (D). Immunoblot analyses of Hes1 levels in three different samples from control, APC−/−, RBP−/− and APC−/−RBP−/− mice (day 5) (E). β-Actin levels were used for normalisation. Alcian blue staining for control, APC−/−, RBP−/− and APC−/−RBP−/− mice (F). Quantitative RT–PCR analysis of Muc2, Gob4 and Gob5 mRNA levels in control, APC−/−, RBP−/− and APC−/−RBP−/− mice (day 5) (G). For statistical analyses mutant mice were compared with control mice. Scale bars, 100 μm.
We checked that the Notch pathway was truly inactivated in the intestine of the double-mutant mice, by showing that efficient recombination of the floxed RBP-J alleles had occurred after tamoxifen injection (figure 5C). We also analysed the expression of Notch target genes. Notch downregulates Atoh1 and the cyclin-dependent kinase inhibitors, p27kip1 and p57kip2.6 15 Quantitative RT–PCR experiments showed the level of expression of the Atoh1, p27kip1 and p57kip2 genes to be specifically increased in double-mutant mice (figure 5D). The persistence of Hes1 expression in the double-mutant mice is consistent with the role of Wnt/β-catenin signalling activation in the regulation of Hes1 expression (figure 5E,4E). Moreover, this underlines the complex regulation of Atoh1 by Hes1 and suggests that other factors are present to remove the repression of Atoh1 expression following RBP deletion. Our findings confirmed that the recombined RBP-J allele was a true null allele and that inhibition of the Notch/RBP-J pathway in the double-mutant mice was effective.
Numerous reports have indicated that the inhibition of Notch signalling leads to forced differentiation into secretory lineages, due to Atoh1 overexpression, resulting in the large-scale conversion of progenitor cells into postmitotic goblet cells.7 15 Apc loss has also been shown to lead to an amplification of multipotent progenitor cells associated with a defect in goblet cell differentiation.2 3 16 We investigated whether the RBP-J ablation associated with Atoh1 mRNA upregulation could rescue the differentiation defects resulting from Apc loss in the intestine. Histological analyses showed that this was not the case. H&E and Alcian blue staining revealed that double-mutant animals displayed an abnormal depletion of mature goblet cells similar to that observed in the Apc-null mice (figure 5B,F). We carefully examined the expression of several markers specific to goblet cells (Muc2, Gob4 and Gob5), comparing the results with those for RBP single-mutant mice (RBP−/− mice). Six days after tamoxifen injection, quantitative RT–PCR experiments showed an increase in their level of expression specifically in the RBP−/− mice (figure 5G). Our results therefore suggest that in the context of acute Wnt/β-catenin activation, Notch inhibition cannot direct terminal differentiation into goblet cells.
We then analysed the effect of RBP-J gene deletion on the cell proliferation defects induced by acute Apc loss. Histological analysis showed that the morphological architecture of the small intestine of double-mutant mice was indistinguishable from that of Apc-null mice (figure 5B). Gross changes associated with Apc deficiency, such as enlarged and aberrant crypts, were observed in the double-mutant mice. We further investigated proliferation status in the double-mutant and Apc-null mice. We assessed levels of bromodeoxyuridine (BrdU) incorporation, and staining for phosphophorylated histone H3 and Ki-67 in double-mutant mice and Apc-null mice, 6 days after tamoxifen injections (figure 6A,B). Ki-67 staining and the numbers of cells in the S and M phases were similar in Apc mutant mice and double-mutant mice. This suggests that RBP-J loss neither delays nor directly affects the cell cycle in the context of Apc loss. Consistent with this finding, all the cell cycle regulators affected by Apc loss were similarly deregulated in the double-mutant intestine (figure 6C). The increase in p27kip1 and p57kip2 levels (figure 5D) resulting from Notch inhibition could not overcome the unrestricted proliferation induced by the aberrant activation of Wnt/β-catenin signalling.

Loss of RBP-J does not affect the increase in cell proliferation in Apc-deficient cells. Analyses of cell proliferation by immunohistochemistry (A). Bromodeoxyuridine (BrdU), phosphorylated histone H3 (pH3) and Ki-67 staining was performed on control, APC−/− and APC−/−RBP−/− mice, 5 days after tamoxifen (Tam) injection. Quantification of BrdU- and pH3-positive cells per crypt in control, APC−/− and APC−/−RBP−/− mice (B). Scale bars, 100 μm. Quantitative reverse transcription–PCR (RT–PCR) analyses of mRNA for various cell cycle components in control, APC−/− and APC−/−RBP−/− mice (day 5). For statistical analyses, mutant mice were compared with control mice (C).
As observed for the proliferation defect, Notch inhibition due to RBP-J loss had no effect on the apoptotic response to Apc loss in the intestine (figure 7A).

Loss of RBP-J does not affect the stem cell amplification or apoptosis induced by Apc deficiency. Identification of apoptotic cells in control, APC−/− and APC−/−RBP−/− mice, 5 days after tamoxifen (Tam) injection, by the deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) method (A). Scale bars, 100 μm. Quantitative reverse transcription–PCR (RT–PCR) analyses of mRNA levels for stem cell markers in control, APC−/− and APC−/−RBP−/− mice (day 5). For statistical analyses, mutant mice were compared with control mice (B).
Finally, we examined the crypt stem cell population, as these cells have been clearly identified as the initial source of intestinal cancer.17 We used quantitative RT–PCR to analyse the expression of several stem cells markers, such as Lgr5, Ascl2, Musashi-1 and Sox9. No difference in stem cell amplification was observed between Apc-null and double-mutant mice (figure 7B).
The inhibition of Notch/RBP-J signalling therefore had no major impact on the early and severe phenotypes induced by Apc loss in the intestine. This suggests that the Notch pathway is dispensable, at least for the initiation of Wnt/β-catenin-induced intestinal tumorigenesis.
Post-transcriptional downregulation of Atoh1 by the activation of β-catenin signalling
Next, we searched for a mechanism which could link the absence of an integrative effect between the Notch inhibition and the Wnt activation in the initiation of intestinal tumorigenesis. One possible explanation is that acute Wnt/β-catenin signalling could modulate a downstream component of Notch. Importantly, it was shown by Watanabe's group that Atoh1 protein is degraded by the Wnt/β-catenin signalling in CRC cell lines.18 19 They showed that Atoh1 protein is targeted by proteasome-mediated degradation via its phosphorylation by glycogen synthase kinase 3β (GSK3β). This was an attractive explanation to support our data. However, in this study, the mechanism of Atoh1 expression stabilisation was based on a forced expression of an Atoh1-tag plasmid. In the current study, we pursued the analysis of Atoh1 expression in three CRC cell lines by analysing the effect of GSK3β or proteasome inhibitors on the endogeneous Atoh1 protein level. First, we confirmed that a forced expression of Tag-Atoh1-HA-tag plasmid was decreased by the oncogenic activation of β-catenin signalling in HEK-293T cells (figure 8A). We thus analysed the activity of the endogenous Atoh1 after treatment with GSK3β or proteasome inhibitors (AR-A014418 and MG132) in SW480, TC7 and HT29 colon cancer cells, three cell lines displaying constitutive activation of β-catenin signalling. The lack of an efficient Atoh1 antibody led us to test this hypothesis with a reporter construct relaying Atoh1 activity. In previous studies, Atoh1 was reported to act in a self-regulatory manner, by binding to an enhancer region containing an E-box site conserved between mice and humans and located in the 3′-untranslated sequence.20 We used a plasmid encoding luciferase under the control of the Atoh1 enhancer–promoter regulatory sequences (Enh-Atoh1-Luc) for transfection analyses. We found that the treatment of SW480, HT29 and TC7 cells with GSK3β or proteasome inhibitors increased Atoh1-mediated transcription (figure 8B). We then analysed the effect of GSK3β and proteasome inhibitors on goblet cell differentiation. Previous studies have shown a direct transactivation of the Muc2 promoter by Atoh1.21 As expected, treatment with GSK3β or proteasome inhibitors increased the activity of the Muc2 promoter–luciferase reporter construct (Muc2-Luc) in all CRC cell lines tested (figure 8C).

β-Catenin signalling controls Atoh1 expression post-transcriptionally. Co-transfections of HEK-293T cells with various amounts of plasmid encoding Atoh1-haemagglutinin (HA) (0, 50 and 150 ng) and an oncogenic mutant form of β-catenin (0 and 500 ng). Atho1 protein levels were analysed by immunoblotting of the HA tag and quantified by Genetool from SynGene software (A). β-Actin levels were used for normalisation. Analyses of Atho1 and Muc2 transcriptional activity in SW480, TC7 and HT-29 cells by transfection with plasmids encoding luciferase under the control of the Atoh1 (Enh-Atoh1-Luc) or Muc2 regulatory sequences (Muc2-Luc). Cells were treated with glycogen synthase kinase 3 β (GSK3β) inhibitor (AR-A014418) for 48 h or with proteasome inhibitor (MG132) for 18 h before the quantification of luciferase activity (B).
In accordance with the observations of Watanabe's group, these results suggest that the aberrant activation of β-catenin signalling leads to Atoh1 degradation in CRC cell lines and that GSK3β or proteasome inhibitors can force the expression of Atoh1 required to drive goblet cell differentiation. They indicate that Atoh1 stabilisation is a critical event in the conversion of β-catenin-activated intestinal epithelial cells into goblet cells.
Atoh1 loss enhances the tumour formation induced by Apc loss
Loss of differentiation is frequently observed during cancer progression and then master regulators of differentiation may be key regulators of tumour formation. In this light, a previous study indicated that mosaic deletion of Atoh1 in the colon increased the number of polyps that developed in ApcMin/+ mice of 5 months of age.22 That study utilised a constitutive Cre transgene driven by the intestine-specific fatty acid-binding protein promoter.23 Here, we pursued this study by using the Villin Cre transgene inducible by tamoxifen (VilCreERT2) which is highly efficient throughout the small and the large intestinal epithelium, and we have analysed the early effect of Atoh1 deletion in the context of heterozygous Apc deletion. For this, we generated double-mutant mice (Atoh1lox/lox; Apclox/+ VilCreERT2) and compared the intestinal lesions developed in these mice with those developed in control mice (Apclox/+ VilCreERT2) 3 weeks after tamoxifen injection (Apc+/–Atoh1−/− vs Apc+/–).23 We checked, by PCR analysis, that tamoxifen injections induced efficient recombination of the floxed Atoh1 and Apc alleles (data not shown). As expected, Atoh1 deletion led to complete ablation of the secretory lineages in both the small and large intestine. Lysosyme and Alcian blue staining indicated a complete loss of Paneth cells and goblet cells throughout the intestine (figure 9A). In addition, all Apc+/−Atoh1−/− mice developed a large number of early adenomas, within 3 weeks, in both the small and large intestine, whereas only rare lesions were observed in Apc+/− mice (figure 9B,C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Loss of Atoh1 enhances the tumour formation induced by Apc loss. Alcian blue staining and immunostaining for lysosyme in Apc+/− and Apc+/−Atoh1−/− mice (A). Mean number of lesions per animal in the small intestine and colon of Apc+/− (n=5) and Apc+/−Atoh1−/− (n=7) mice (B). H&E staining and immunostaining for β-catenin in Apc+/−Atoh1−/− mice (C).
Thus, the absence of Atoh1 rapidly promotes the development of intestinal tumours induced by Apc loss. Atoh1 acts as a master gene that instructs cell fate commitment in secretory lineages and acts as a brake on oncogenic process throughout the whole intestinal tract.
Discussion
Several recent studies have suggested that the Wnt and Notch pathways cooperate to trigger intestinal tumorigenesis.7 9 12 However, the molecular mechanism involved in this cross-talk remains unclear. We have shown that Notch activation, as reflected by overproduction of the Hes1 and Notch ligands and receptors, is a frequent and early event in Wnt-induced intestinal tumorigenesis, and that this activation is maintained throughout tumorigenesis and downstream from the Wnt/β-catenin cascade. We show that Hes1 overexpression in intestinal cancer results from two complementary processes controlled by β-catenin. One is the β-catenin-mediated upregulation of the ligands and receptors of the Notch pathway, as previously described.9 12 24 However, among all the Notch ligands and receptors tested, only the expression of Jag1 appears directly affected by β-catenin activation. The upregulation of the other members appears to be an indirect consequence of the activation of the β-catenin signalling. The second process is a direct increase in Hes1 promoter activity mediated by β-catenin/Tcf signalling. Indeed, although Hes1 is a genuine Notch effector, it may also be activated by other signalling pathways, such as those of Wnt, Sonic Hedgehog, IκBα and Ras/MAPK.25–28
Altogether, we and others showed a complex integration of the Wnt and Notch pathways in intestinal epithelial cells. During intestine development, it has been shown that the effects of Notch signalling on intestinal cell proliferation are dependent on Wnt. This epistatic study indicates that Notch acts upstream of Wnt. However, in adult intestine, during Apc-driven tumorigenesis, our data indicate that activation of Notch acts downstream of Wnt. This statement is in accordance with other published data.12 29 Altogether, it appears that the interaction between the Wnt and the Notch pathways is complex and appears to be context dependent.
We analysed the role of Notch in tumorigenesis, by assessing the effect of blocking Notch signalling in cells in which β-catenin was deregulated, mimicking the early stage of tumorigenesis. We used conditional targeting methods to delete the Apc and RBP-J genes simultaneously in the intestinal epithelium of adult mice. As the treatment of ApcMin adenomas with γ-secretase inhibitors led to the conversion of some adenoma cells to goblet cells,7 we expected to alter the initiation of tumour development induced by acute Apc loss. However, Notch inhibition did not rescue the severe phenotype caused by Apc loss in the intestine. No commitment to goblet cell lineage differentiation was observed, and unrestricted proliferation, with enlarged, highly proliferative crypts and stem cell amplification was seen in all cases, regardless of Notch status. Based on published data, one possible explanation is that acute Wnt/β-catenin signalling could modulate expression of Atoh1, a downstream component of Notch.18 19 Using CRC cell lines, we showed that endogenous Atoh1 protein levels were downregulated by Wnt/β-catenin signalling. The stabilisation of Atoh1 protein by GSK3β and proteasome inhibitors allowed the expression of the goblet cell marker. Conversely, we showed that genetic ablation of Atoh1 dramatically accelerated in 3 weeks the emergence of Apc-mediated adenomas throughout the whole small and large intestine (results of this study,22). Consistent with the tumour suppressor role of Atoh1 in CRCs, recent studies have reported the epigenetic and genetic silencing of the Atoh1 gene in human CRC.22 These results highlight a critical role for Atoh1 in the oncogenic effect of β-catenin signalling in intestinal tumorigenesis.
Our data contrast with those of a study reporting the conversion of adenoma cells into goblet cells after the treatment of ApcMin mice with γ-secretase inhibitors.7 However, conversion to goblet cells was nonetheless relatively rare. Almost 50% of adenomas showed no conversion of tumours cell and only 28% displayed the conversion of 1–10% of tumour cells into goblet cells.7 There may be several reasons for the discrepancies between these two studies. As Notch inactivation was achieved by deletion of the RBP gene or with γ-secretase inhibitors, we cannot exclude the possibility of an RBP-independent effect of Notch.30 However, it is more likely that the different effects of Notch inhibition on the Apc-mediated phenotype are linked to different β-catenin activation thresholds. Indeed, a dose-dependent effect of β-catenin has already been described.31 32 Based on the expression profile of the β-catenin target gene c-myc that we have previously described,3 10 it appears that the level of activation of Wnt/β-catenin signalling differs between the two mouse models. Strong and homogeneous β-catenin activation probably leads to Atoh1 destabilisation in the presence of a conditional homozygous deletion of Apc, whereas the heterogeneous β-catenin activation in ApcMin mice may allow Notch inhibition by maintaining Atoh1 levels above a threshold sufficient to induce goblet differentiation, in a subset of cells. A recent study showed that the complete deletion of Atoh1 abolished the effects of γ-secretase inhibitors on cancerous intestinal cells.33
Our data highlight the complex interplay between the Wnt and Notch signalling pathways during intestinal tumorigenesis. Two main Notch effectors are controlled in opposite ways by the Wnt/β-catenin pathway. Hes1 is transcriptionally upregulated, whereas Atoh1 is post-transcriptionally downregulated. This downregulation is critical to the oncogenic outcome mediated by β-catenin, because it prevents the differentiation of cancer cells. Our findings have important clinical implications, as they call into question the utility of γ-secretase inhibitors as a treatment for CRC and suggest that the stabilisation of Atoh1 may be of therapeutic importance.
Acknowledgments
We thank B.Z. Stanger and T. Sudo for Hes1 antibodies, R. Kageyama for the Hes1 reporter gene, J. Johnson for Enh-Atoh1-Luc, B. Radenen for technical assistance, the animal facility staff and team members for helpful discussions, and in particular Dr Jean-Pierre Couty and Marie Anson for their invaluable help.
References
Supplementary materials
online only appendix
Files in this Data Supplement:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Funding This work was supported by grants from INCa, ARC, the Ligue Nationale Contre le Cancer, Inserm and the CNRS.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Local Ethical Commitee Paris Cochin Hospital 2002 (Paris, France).
Provenance and peer review Not commissioned; externally peer reviewed.